Page 11 of 25
PH1.7-9 | PH1.7-9 | Pharmacodynamics, Prototype Drug Effects and Rational Combinations — SDL Guide — SDL Guide
Learning Objectives
- Describe the major mechanisms of drug action: receptor-based (agonism, antagonism) and non-receptor-based (enzyme inhibition, ion channel modulation, physicochemical effects).
- Distinguish full agonist, partial agonist, inverse agonist, and competitive/non-competitive antagonism using dose-response curves.
- Explain dose-response curve parameters (Emax, EC50, potency, efficacy) and the therapeutic index.
- Demonstrate the PD principles of key prototype drugs (morphine, atropine, aspirin, adrenaline, propranolol).
- Define and classify drug combination outcomes: synergism (additive, potentiation), antagonism (pharmacological, chemical, physiological), and apply PK-PD rationale to select rational combinations.
INSTRUCTIONS
Pharmacodynamics answers the most fundamental question in pharmacology: how does a drug produce a biological effect? The answer determines everything downstream — which organs are affected, what the dose-response relationship looks like, how long the effect lasts, why some effects are desirable and others are adverse, and why combining two drugs can produce additive benefit, dangerous synergistic toxicity, or complete antagonism. Understanding pharmacodynamics at the mechanistic level converts drug effects from a catalogue of facts to memorise into a predictable system to reason from.
References
- Tripathi KD. Essentials of Medical Pharmacology, 8th ed., Ch 3-4 (textbook)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, 13th ed., Ch 3 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 70-year-old man is brought in unconscious after a suspected opioid overdose. He has pinpoint pupils, a respiratory rate of 4 breaths/minute, and a GCS of 6. You draw up naloxone and administer 0.4 mg IV. Within 90 seconds, he opens his eyes, his respiratory rate rises to 16 breaths/minute, and he asks where he is. Twenty minutes later he becomes drowsy again — the naloxone has worn off but the long-acting opioid he took is still at the receptor. You need a second dose. Everything that just happened — the dramatic reversal of opioid effects, the competitive displacement of opioid from the mu receptor, the return of toxicity when the shorter-acting competitive antagonist wears off while the agonist remains — is pharmacodynamics in clinical action.
WHY THIS MATTERS
Every prescribing decision you make involves predicting the pharmacodynamic consequences of a drug in a specific patient. Will this beta-blocker lower heart rate? Will this antihistamine cause sedation? Can I combine a diuretic with an ACE inhibitor for better blood pressure control, and if so why? Will mixing a benzodiazepine with opioids dangerously potentiate respiratory depression? These questions are unanswerable from drug names alone — they require understanding the mechanistic basis of drug action, the shape of the dose-response relationship, and the principles governing drug combinations. This SDL provides that mechanistic foundation.
RECALL
From Year-1 biochemistry and physiology, you know that receptors are protein molecules (or nucleic acids in some cases) that specifically bind ligands — endogenous hormones, neurotransmitters, or exogenous drugs — and transduce that binding into a cellular signal. You have encountered G protein-coupled receptors (adenylyl cyclase and phospholipase C second-messenger pathways), ligand-gated ion channels (nicotinic acetylcholine receptor), enzyme-linked receptors (insulin receptor tyrosine kinase), and nuclear receptors (steroid hormone receptors). These receptor classes are the pharmacodynamic targets of the majority of clinically used drugs.
How Drugs Produce Their Effects: The Receptor Hypothesis
The receptor hypothesis — the idea that drugs produce their effects by combining with specific biological macromolecules called receptors — is the central organising principle of pharmacodynamics. It was first articulated by John Newport Langley (1878, studying the antagonism between nicotine and curare at the neuromuscular junction) and independently by Paul Ehrlich (1908, proposing that drugs must 'bind' to cellular constituents to produce effects — his famous metaphor: the drug is the bullet, the receptor is the target). Without the receptor concept, the relationship between drug structure and pharmacological activity would be an uninterpretable empirical catalogue.
The receptor hypothesis predicts several properties that are now confirmed as foundational facts of pharmacology. First, drug effects are selective — a drug acts only on cells that express the receptor it binds to. Morphine analgesia is selective for opioid receptors; it does not block sodium channels or activate adrenergic receptors at therapeutic doses. Second, drug effects are saturable — there are a finite number of receptor molecules, and once all are occupied, increasing drug concentration adds no further effect (the Emax plateau of the dose-response curve). Third, drug effects are reversible — most drug-receptor interactions involve non-covalent bonds (ionic, hydrogen, van der Waals) that can be broken by dissociation of the drug or competition from an antagonist, as in the naloxone reversal seen in the hook.
Four major receptor superfamilies account for the targets of the vast majority of therapeutic drugs: G protein-coupled receptors (GPCRs) — the largest family, linked to adenylyl cyclase (cAMP) or phospholipase C (IP3/DAG/Ca2+) second messengers; examples: beta-adrenergic (cAMP), muscarinic (IP3), opioid (inhibitory Gi). Ligand-gated ion channels (LGICs) — open or close in response to ligand binding with millisecond kinetics; examples: nicotinic ACh receptor (Na+/K+), GABA-A receptor (Cl−). Enzyme-linked receptors — have intrinsic enzyme activity activated by ligand binding; examples: insulin receptor (tyrosine kinase), natriuretic peptide receptor (guanylyl cyclase). Nuclear/intracellular receptors — ligand diffuses into the cell and binds cytoplasmic or nuclear receptors that then act as transcription factors; examples: glucocorticoid receptor, thyroid hormone receptor. Drug effects are slower onset (hours, because protein synthesis is required) but more prolonged.
Therapeutic Goals: Selectivity, Efficacy, and Safety
The ideal drug is perfectly selective (acts only on the target receptor in the target tissue), maximally efficacious (produces the full desired effect), and perfectly safe (no toxicity at any clinically needed dose). No real drug meets all three criteria, but understanding the quantitative parameters that describe how close a drug comes to this ideal is essential for rational drug selection and dosing.
Dose-response curves describe the relationship between drug dose (or concentration) and the magnitude of biological effect. The sigmoid dose-response curve (log dose on x-axis, effect as percentage of maximum on y-axis) has two parameters of central importance:
- Emax (maximal effect): the ceiling response — the maximum effect achievable regardless of further dose increase. Emax is a measure of efficacy (intrinsic activity): how well the drug can activate the receptor even when all receptors are occupied.
- EC50 (or ED50 for in vivo): the concentration (or dose) producing 50% of Emax. EC50 is a measure of potency: how little drug is needed to produce a given effect. A drug with lower EC50 is more potent — it achieves a given effect at a lower dose. Potency and efficacy are independent; a drug can be potent but have low efficacy (partial agonist) or highly efficacious but require a large dose (low potency).
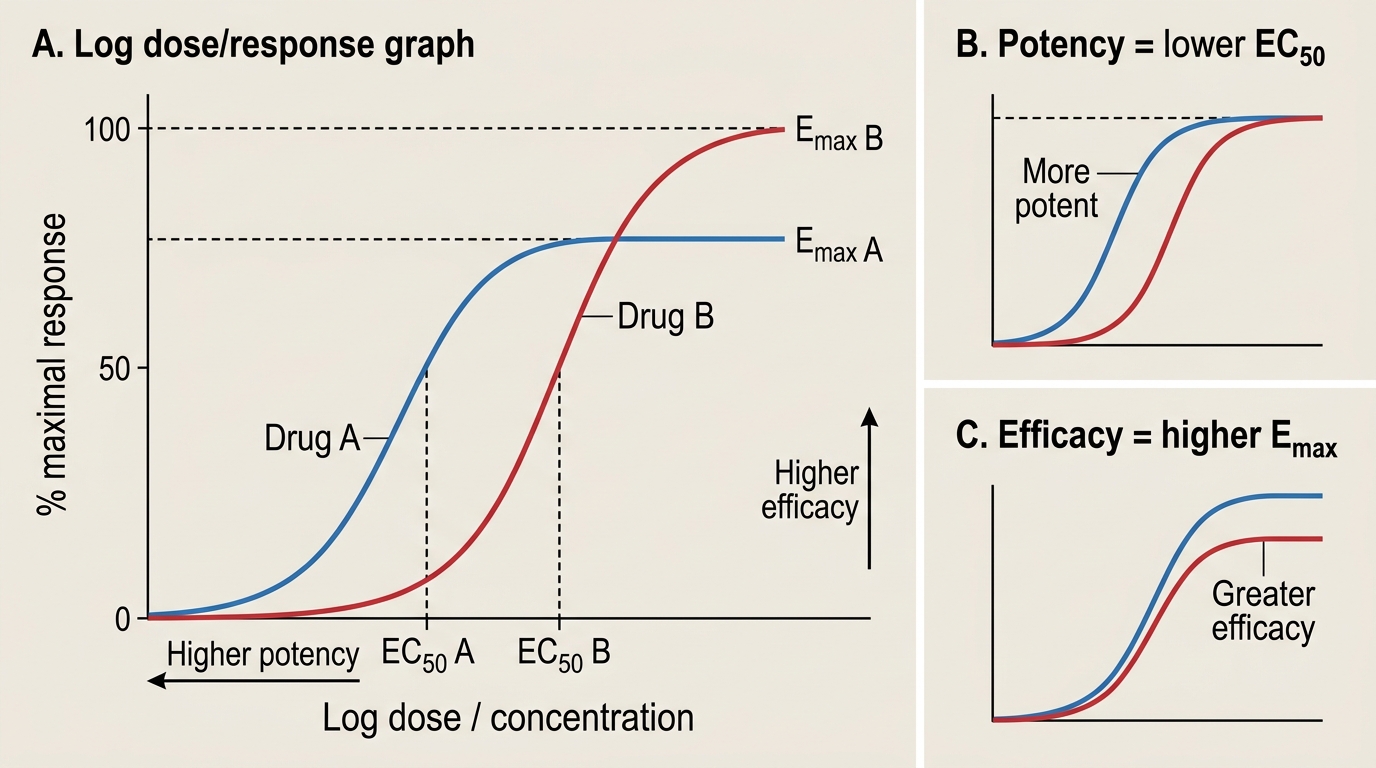
Potency and Efficacy on Dose-Response Curves
The therapeutic index (TI) quantifies the margin between therapeutic and toxic doses in a population: TI = TD50 / ED50, where TD50 is the dose producing toxicity in 50% of the population and ED50 is the dose producing the desired effect in 50%. A wide TI (e.g., penicillin TI >> 100) means large doses can be given safely above the therapeutic threshold. A narrow TI (e.g., digoxin TI ≈ 2, phenytoin TI ≈ 2, lithium TI ≈ 2–3) means the toxic dose is only slightly above the effective dose — small deviations from the intended dose can cause toxicity, necessitating therapeutic drug monitoring (TDM).
Selectivity refers to the degree to which a drug acts on one receptor type or tissue relative to others. No drug is perfectly selective at high doses — as concentration rises, drugs begin to interact with structurally similar off-target receptors, producing unwanted effects. Beta-1 selective adrenergic blockers (metoprolol, atenolol) are preferred over non-selective beta-blockers (propranolol) in asthmatic patients because at therapeutic doses they spare beta-2 receptors in the bronchi — though selectivity diminishes at high doses.
Receptor-Based Mechanisms: Agonists, Antagonists, and Receptor Types
Drugs that interact with receptors can be classified by whether they activate the receptor (agonists) or prevent activation (antagonists), and by the degree of activation they produce relative to the endogenous ligand.
A full agonist binds the receptor and produces the maximal possible response — it has both high affinity (binds readily) and high intrinsic efficacy (activates the receptor fully). Morphine is a full agonist at the mu-opioid receptor: at sufficient doses it produces 100% of the receptor's possible analgesic, respiratory depressant, and euphoric effects.
A partial agonist binds the receptor but produces a submaximal response even when all receptors are occupied — it has high affinity but low intrinsic efficacy. Buprenorphine is a partial agonist at the mu-opioid receptor: it produces good analgesia but cannot produce as much respiratory depression as morphine even at saturating doses, making it safer in overdose. Importantly, because buprenorphine has higher receptor affinity than morphine, it can displace morphine from the receptor and precipitate withdrawal in opioid-dependent patients — a counter-intuitive but pharmacologically predictable effect.
An inverse agonist binds the receptor and produces an effect opposite to that of the agonist — it has intrinsic negative efficacy. This is different from an antagonist, which simply blocks without activating. Beta-carbolines are inverse agonists at the GABA-A benzodiazepine site: they produce anxiogenic and convulsant effects, the opposite of benzodiazepine agonists.
Antagonists block agonist action without producing their own effect. A competitive reversible antagonist competes with the agonist for the same binding site. It shifts the dose-response curve rightward — a higher agonist concentration is needed to produce the same effect, but the same Emax is still achievable. Naloxone, atropine, and propranolol are competitive reversible antagonists. A competitive irreversible antagonist (e.g., phenoxybenzamine at alpha-adrenergic receptors — covalent bond via alkylation) cannot be displaced by increasing agonist; it reduces Emax. A non-competitive antagonist binds at a different (allosteric) site, reducing receptor responsiveness without competing for the agonist binding site; it also reduces Emax but does not shift EC50.
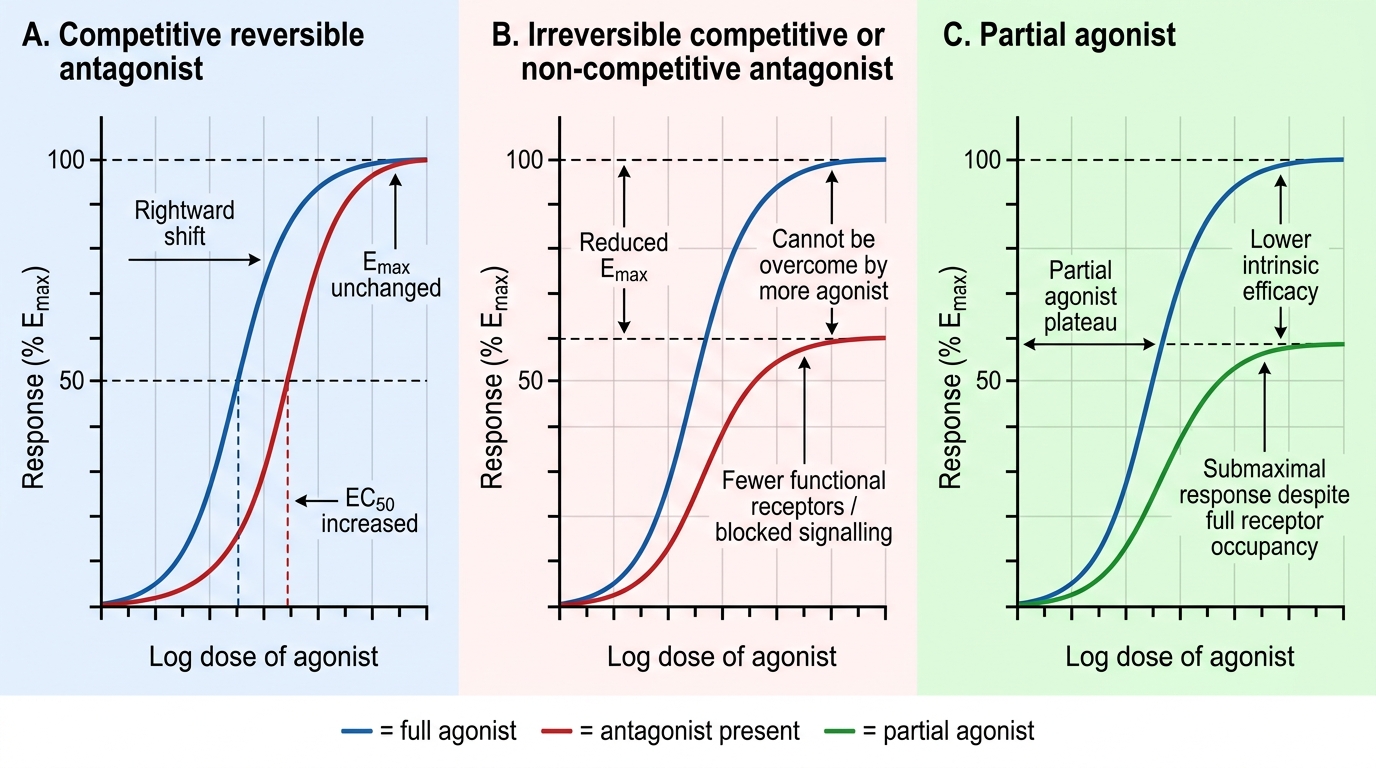
Dose-Response Curve Changes with Antagonists and Partial Agonists
Receptor regulation occurs with prolonged agonist or antagonist exposure. Prolonged agonist exposure causes down-regulation (reduced receptor number and/or responsiveness) and desensitisation (uncoupling of receptor from its effector) — the pharmacological basis of drug tolerance. Prolonged antagonist exposure causes up-regulation (increased receptor number), which explains why abrupt withdrawal of a beta-blocker can cause rebound tachycardia and angina — sudden removal of antagonist unmasks supersensitive, up-regulated beta-adrenergic receptors.
SELF-CHECK
Naloxone reverses opioid toxicity by competing with opioids at the mu receptor. After reversal, you observe the patient becomes drowsy again 30 minutes later. Which pharmacokinetic-pharmacodynamic explanation is correct?
A. Naloxone has induced tolerance at the mu receptor, requiring more opioid to be effective.
B. Naloxone (t½ ~60–90 min) has been eliminated faster than the longer-acting opioid still bound to the receptor; opioid effect returns as naloxone concentration falls below the competitive threshold.
C. The opioid has undergone biotransformation to a more potent active metabolite during the treatment.
D. Up-regulation of mu receptors occurred within 30 minutes of naloxone administration.
Reveal Answer
Answer: B. Naloxone (t½ ~60–90 min) has been eliminated faster than the longer-acting opioid still bound to the receptor; opioid effect returns as naloxone concentration falls below the competitive threshold.
Naloxone is a competitive reversible antagonist with a short half-life (~60–90 minutes IV). Many opioids (e.g., methadone, sustained-release oxycodone, buprenorphine) have much longer half-lives. As naloxone plasma concentration falls, competitive displacement weakens, opioid concentration at the receptor rises relative to naloxone, and opioid agonism re-emerges. This is why patients given naloxone must be observed for at least 2–4 hours, or given a naloxone infusion, after reversal of long-acting opioids.