Page 20 of 33
PH4.10 | PH4.10 | Antiarrhythmic Drugs — SDL Guide
Learning Objectives
- Explain the Vaughan-Williams classification of antiarrhythmic drugs and describe the mechanism of each class.
- Describe the pharmacokinetics, pharmacodynamics, therapeutic uses, and adverse drug reactions of the major antiarrhythmic agents.
- Devise a pharmacological management plan for supraventricular arrhythmias (SVT, AF), ventricular arrhythmias (VT), cardiac arrest (VF), and AF in Wolff-Parkinson-White syndrome.
- Identify drugs that are pro-arrhythmic and explain the mechanism by which they cause QT prolongation and torsades de pointes.
INSTRUCTIONS
Antiarrhythmic pharmacology is one of the most intellectually demanding areas of pharmacology — it requires understanding cardiac electrophysiology, drug mechanisms at the ion-channel level, and the paradox of pro-arrhythmia (where the very drugs intended to treat arrhythmias can cause worse arrhythmias). The Vaughan-Williams classification provides a framework, but clinical decision-making requires knowing which drug is appropriate for which arrhythmia, and critically, which drugs to avoid in specific contexts such as structural heart disease, WPW syndrome, and QT prolongation states.
References
- Tripathi KD. Essentials of Medical Pharmacology, 9th ed., Ch. 36 (Antiarrhythmic Drugs) (textbook)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, 14th ed., Ch. 29 (Antiarrhythmic Drugs) (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 34-year-old woman is brought to the emergency department with palpitations and dizziness. ECG shows a narrow-complex regular tachycardia at 180 bpm with no visible P waves — consistent with AV nodal re-entrant tachycardia (AVNRT). Her BP is 110/70. While the nurse prepares adenosine, you notice that the ECG in the medical record from 2 years ago shows a delta wave and short PR interval — findings consistent with Wolff-Parkinson-White (WPW) syndrome. One of your colleagues immediately hands you verapamil for SVT. Should you give it? What will happen if you do? And which drug should you actually use — and why?
WHY THIS MATTERS
Arrhythmias range from benign palpitations to life-threatening cardiac arrest. Every acute care physician, emergency doctor, cardiologist, and internist must be able to make split-second drug decisions for arrhythmias — decisions where the wrong drug can convert an atrial arrhythmia into ventricular fibrillation (as in WPW + verapamil) or worsen underlying disease (as in flecainide post-MI). The antiarrhythmic drugs are one of the few drug classes where a prescribing error based on misidentified arrhythmia type can cause immediate death. Understanding the electrophysiological basis of these drugs — not just their names and classes — equips you to make correct decisions under pressure.
RECALL
Recall from physiology (PY) the cardiac action potential:
- Phase 0 — rapid depolarisation: fast Na channels (INa) open → Na influx → upstroke (non-nodal tissue); slow Ca channels (ICa-L) responsible in SA/AV node (nodal tissue lacks phase 0 Na current).
- Phase 1 — early repolarisation: transient K+ outward current.
- Phase 2 — plateau: L-type Ca channels (ICa-L) remain open; K channels begin to open; Ca influx maintains the plateau — the basis of cardiac contraction coupling.
- Phase 3 — rapid repolarisation: K channels (IKr, IKs) fully open → K efflux → repolarisation back to resting potential.
- Phase 4 — diastolic depolarisation (pacemaker cells only): If (funny current), small Na influx → slow spontaneous depolarisation until threshold → next action potential (automaticity).
- AV node: slow-channel tissue (depends on ICa-L, not INa) → responsible for the PR interval delay; can be slowed by Class II (beta-blockers), Class IV (verapamil, diltiazem), adenosine, and digoxin.
- Accessory pathway (WPW): a bypassing tract connecting atria to ventricles, bypassing the AV node. In WPW, rapid atrial impulses (e.g., in AF) can conduct rapidly via the accessory pathway → ventricular pre-excitation → risk of VF.
Cardiac Action Potential and Arrhythmia Mechanisms
Arrhythmias arise from one of three electrophysiological mechanisms: abnormal automaticity, triggered activity, or re-entry — and rational antiarrhythmic drug selection requires understanding which mechanism underlies the arrhythmia being treated.
Automaticity refers to the capacity of pacemaker cells (SA node, AV node, and latent pacemakers in His-Purkinje cells) to spontaneously depolarise during phase 4. Abnormal automaticity occurs when non-pacemaker cells develop spontaneous depolarisation — typically due to ischaemia, hypokalaemia, or catecholamine excess. Beta-blockers (Class II) and direct SA node rate-reducers (ivabradine) target this mechanism.
Triggered activity involves abnormal depolarisations that occur in the wake of a preceding action potential — either during repolarisation (early after-depolarisations, EADs — caused by drugs that prolong the QT interval: Class Ia/Class III agents, especially when combined with hypokalaemia) or after repolarisation (delayed after-depolarisations, DADs — caused by intracellular Ca overload, as in digoxin toxicity or catecholamine excess). EADs are the electrophysiological basis of torsades de pointes (TdP) — a potentially fatal polymorphic ventricular tachycardia.
Re-entry is the commonest mechanism for sustained tachyarrhythmias — including AVNRT (AV nodal re-entrant tachycardia), atrial flutter, and many forms of VT. It requires two pathways with different conduction velocities and refractory periods — one slower and one faster — creating a circular conduction loop that perpetuates itself without re-triggering from the SA node. Drugs that slow conduction (Class I — Na-channel blockers) or increase refractoriness (Class III — K-channel blockers) interrupt re-entry.
The cardiac action potential ion channels that antiarrhythmic drugs target map directly to the Vaughan-Williams classification:
- Class I: fast INa (Phase 0) — blocks conduction velocity
- Class II: catecholamine-β₁ receptor coupling (Phase 4 automaticity)
- Class III: IKr/IKs (Phase 3 repolarisation) — prolongs AP duration + refractoriness
- Class IV: ICa-L (Phase 2 plateau and nodal tissue Phase 0/4)
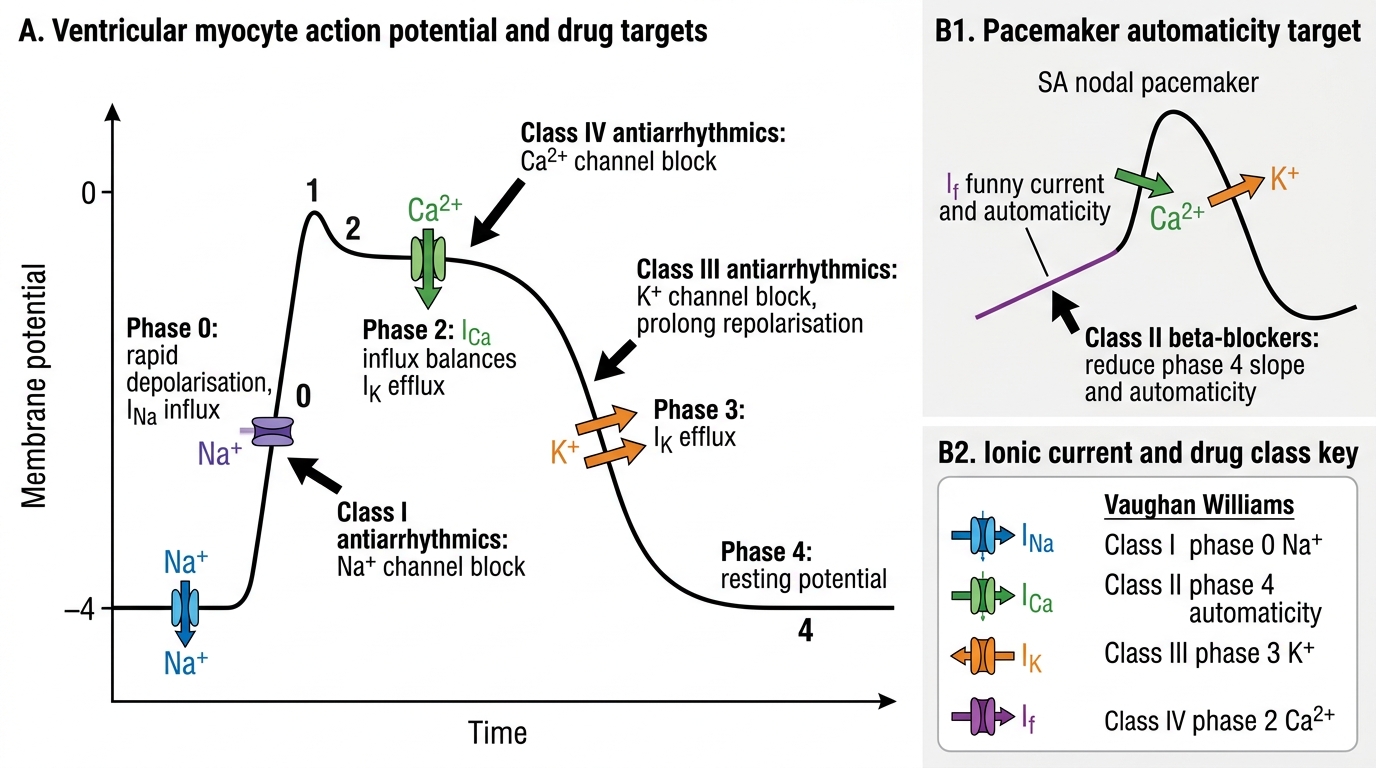
Cardiac Action Potential Phases and Antiarrhythmic Drug Targets
Therapeutic Goals in Arrhythmia Management
The therapeutic goals in arrhythmia management depend fundamentally on whether the arrhythmia is life-threatening (requiring immediate termination and prevention of recurrence) or symptom-causing (requiring rate or rhythm control for quality of life).
Rate control vs rhythm control — the central paradigm in atrial fibrillation management:
- Rate control: slow the ventricular response to AF (target HR <80–100 bpm at rest) using AV-nodal blocking agents (beta-blockers, non-DHP CCBs, digoxin) — without attempting to restore sinus rhythm. The AFFIRM trial showed rate control and rhythm control (antiarrhythmic drugs) produced similar cardiovascular outcomes in most AF patients, with rate control having fewer drug-related adverse events. Rate control is the default strategy for permanent or persistent AF.
- Rhythm control: cardiovert to and maintain sinus rhythm using antiarrhythmic drugs (amiodarone, flecainide, propafenone, sotalol) or electrical cardioversion (DC cardioversion). Rhythm control is preferred in: recent-onset AF (<48h), AF causing haemodynamic compromise, AF with HFrEF where sinus rhythm is strongly preferable, and younger patients (EAST-AFNET-4 trial suggested early rhythm control in selected patients reduces composite cardiovascular events).
Life-threatening arrhythmias (VT/VF): The goal is immediate termination of the arrhythmia to restore cardiac output. In pulseless VT or VF: defibrillation is the definitive treatment; IV amiodarone is the first-line drug adjunct (ACLS algorithm). For haemodynamically stable VT: IV amiodarone or IV lidocaine; electrical cardioversion if drug-refractory. ICD (implantable cardioverter-defibrillator) provides long-term protection against sudden cardiac death — superior to antiarrhythmic drugs for secondary prevention of life-threatening ventricular arrhythmias (AVID trial).
Pro-arrhythmia — the critical limitation of antiarrhythmic drugs: most antiarrhythmic drugs are also pro-arrhythmic under some conditions. The CAST trial demonstrated that Class Ic drugs (flecainide, encainide) increased mortality in post-MI patients with asymptomatic ventricular ectopy — despite successfully suppressing the ectopy. The mechanism: Class Ic drugs depress conduction in partially ischaemic myocardium, creating substrate for re-entry ventricular arrhythmias. This is the principle of use-dependence — some Na-channel blockers block more effectively at faster heart rates, creating paradoxical vulnerability in high-rate states.
The Vaughan-Williams Classification and Beyond
The Vaughan-Williams (VW) classification categorises antiarrhythmic drugs by their primary ion-channel target in cardiac tissue. It is the standard framework in pharmacology and clinical practice, though it has limitations (many drugs have multiple actions; mechanism may not perfectly predict clinical utility).
CLASS I — Sodium-channel blockers: Block fast inward Na current (Phase 0) → slowed conduction velocity (widened QRS). Divided by rate of drug dissociation from the Na channel (recovery kinetics):
- Class Ia (intermediate recovery — quinidine, procainamide, disopyramide): also block K channels → prolong QT; widen QRS; risk of TdP; quinidine also has anti-vagal effects (paradoxical reflex tachycardia). Largely superseded.
- Class Ib (fast recovery — lidocaine, mexiletine, tocainide): selectively block activated/inactivated Na channels in depolarised (ischaemic) tissue; minimal effect on normal tissue; do NOT prolong QT. Lidocaine IV is used for VT in acute MI.
- Class Ic (slow recovery — flecainide, propafenone): most potent Na-channel block; minimal QT prolongation; CAST trial established absolute contraindication in structural heart disease or post-MI (increased mortality via pro-arrhythmia). Reserved for AF with a structurally normal heart.
CLASS II — Beta-adrenergic blockers: Propranolol, metoprolol, atenolol, bisoprolol — reduce sympathetically driven automaticity (↓phase 4 slope in SA/AV node) and slow AV conduction. First-line rate control for AF and SVT; reduce post-MI arrhythmic sudden death.
CLASS III — Potassium-channel blockers: Block IKr (rapid delayed rectifier K current) → ↑action potential duration (APD) → ↑effective refractory period → interrupted re-entry. All prolong QT → TdP risk:
- Amiodarone: unique among Class III agents in having FOUR drug class actions (I+II+III+IV) — a 'pan-channel blocker.' Most effective antiarrhythmic available; used for AF, VT, VF prophylaxis, and cardiac arrest. Longest half-life of any cardiac drug (~40–55 days) → loading dose required; slow onset and offset.
- Sotalol: Class III (IKr) + Class II (beta-blockade); used for AF and VT; QT monitoring mandatory (K and Mg must be normal before use); torsades de pointes risk.
- Dofetilide, ibutilide: pure IKr blockers; AF conversion; hospital-only initiation due to QT monitoring requirement.
CLASS IV — L-type calcium-channel blockers (non-dihydropyridines): Verapamil, diltiazem — block L-type Ca channels in the SA and AV nodes → ↓automaticity + ↓AV conduction velocity. Used for: SVT (terminate or prevent AV-nodal dependent re-entry), AF rate control (IV or oral). Contraindicated with beta-blockers (combined blockade → severe bradycardia/AV block). Contraindicated in WPW + AF — by blocking AV node conduction, they force impulses through the accessory pathway → very rapid ventricular rate → VF.
BEYOND THE VW CLASSIFICATION:
- Adenosine: endogenous nucleoside activating A1 receptors on AV node cells → G-protein-coupled increase in K+ conductance → hyperpolarisation → AV nodal block for ~10–30 seconds. Terminates all AV-node-dependent SVTs within seconds. Very short half-life (~10 seconds). Administered as rapid IV bolus (6 mg, then 12 mg × 2). Side effects: transient chest tightness, flushing, dyspnoea (bronchospasm — caution in severe asthma), transient heart block. Safe in pregnancy.
- Digoxin: vagally-mediated AV nodal slowing (rate control in AF); used when rate control without negative inotropy is needed (or in HFrEF as adjunct).
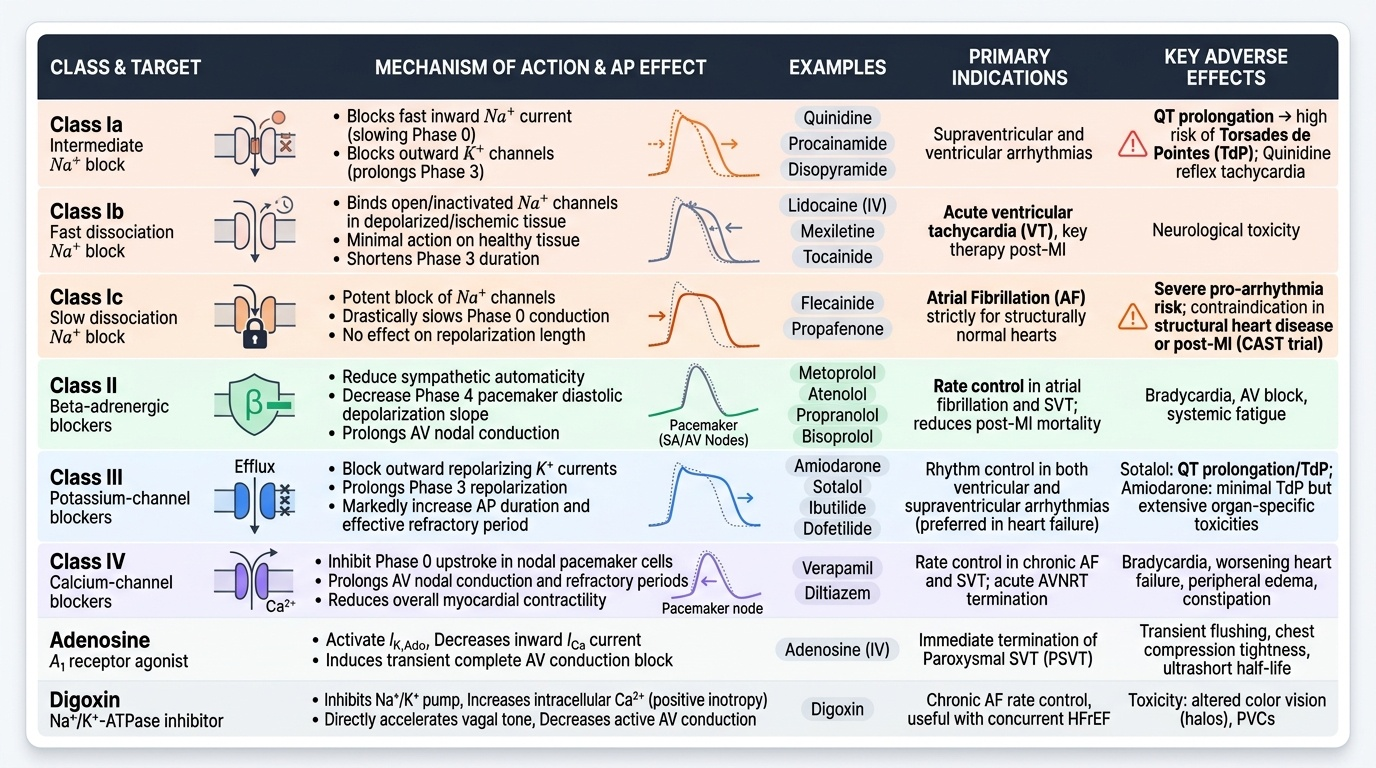
Provided image
SELF-CHECK
Which of the following statements about flecainide (Class Ic antiarrhythmic) is CORRECT?
A. Flecainide can be safely used in patients with post-MI ventricular ectopy to suppress arrhythmias
B. Flecainide is the drug of choice for VT in acute myocardial infarction
C. Flecainide is used for AF in patients with structurally normal hearts but is contraindicated after MI or in LV dysfunction
D. Flecainide prolongs the QT interval and causes torsades de pointes as its primary toxicity
Reveal Answer
Answer: C. Flecainide is used for AF in patients with structurally normal hearts but is contraindicated after MI or in LV dysfunction
Flecainide (Class Ic) is one of the most potent Na-channel blockers available and is effective for AF rhythm control — but only in patients with structurally normal hearts and preserved LV function. The CAST trial (Cardiac Arrhythmia Suppression Trial) demonstrated that flecainide and encainide increased mortality in post-MI patients with asymptomatic ventricular ectopy, despite suppressing the ectopy — due to pro-arrhythmic effects in ischaemic myocardium. Flecainide is therefore CONTRAINDICATED after MI, in HF, or in any structural heart disease. Its primary toxicity is QRS widening (Na-channel block → slowed conduction) rather than QT prolongation (which is mainly a Class Ia and Class III effect).