Page 1 of 33
PH4.11 | PH4.11 | Dyslipidaemia Pharmacotherapy — SDL Guide
Learning Objectives
- Explain the pathophysiological link between dyslipidaemia and atherosclerotic cardiovascular disease (ASCVD).
- Classify lipid-lowering drugs by mechanism and describe the salient pharmacokinetics, pharmacodynamics, therapeutic uses, and adverse drug reactions of each class.
- Enumerate drugs that cause dyslipidaemia and identify their mechanisms.
- Devise a rational pharmacotherapy plan for patients with dyslipidaemia across risk categories including special populations.
INSTRUCTIONS
Dyslipidaemia is the single most modifiable risk factor for ischaemic heart disease — reducing LDL-cholesterol by even 1 mmol/L cuts major cardiovascular events by roughly 22% (Cholesterol Treatment Trialists' meta-analysis). As a prescribing physician you will decide not only which drug to start, but at what intensity, how to monitor, and how to manage statin intolerance in patients who cannot tolerate first-line therapy. This SDL builds the mechanistic foundation and the prescribing framework you will apply at the bedside.
References
- Tripathi KD. Essentials of Medical Pharmacology, 9th ed., Ch. 33 (Hypolipidaemic Drugs) (textbook)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, 14th ed., Ch. 28 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 54-year-old male teacher presents to the medicine outpatient clinic for a routine health check. He is a known hypertensive on amlodipine and has a 15-pack-year smoking history. His fasting lipid profile shows: Total cholesterol 268 mg/dL, LDL-C 182 mg/dL, HDL-C 38 mg/dL, Triglycerides 240 mg/dL. He has no chest pain and his ECG is normal. 'My father died of a heart attack at 58,' he tells you. 'Do I really need to start a tablet just for cholesterol?' He has never taken a lipid-lowering drug. How do you calculate his cardiovascular risk, and what is your prescribing decision?
WHY THIS MATTERS
Cardiovascular disease accounts for approximately one-third of all deaths globally, and dyslipidaemia — principally elevated LDL-cholesterol — is its most tractable risk factor. Statins are among the most widely prescribed and evidence-tested drugs in modern medicine; as an intern and junior resident you will initiate, titrate, and troubleshoot them almost daily. Understanding the full pharmacological landscape — including newer agents such as PCSK9 inhibitors and the obligatory knowledge of which drugs cause dyslipidaemia — is essential for safe, evidence-based cardiovascular prescribing across your entire clinical career.
RECALL
Before proceeding, recall the following from your Year-1 biochemistry and physiology:
- Cholesterol biosynthesis (BI): the pathway runs acetyl-CoA → HMG-CoA → mevalonate (the rate-limiting HMG-CoA reductase step) → farnesyl pyrophosphate → squalene → cholesterol. Statins block this critical step.
- Lipoprotein classification (PY/BI): chylomicrons transport dietary fat; VLDL carries hepatic fat to periphery; IDL is the transition; LDL is the atherogenic end-product; HDL mediates reverse cholesterol transport. LDL-R on hepatocytes mediates LDL clearance — higher receptor expression = lower circulating LDL.
- PCSK9 degrades LDL receptors after their recycling cycle, thereby limiting hepatic LDL clearance; this provides the therapeutic target for PCSK9 inhibitors.
- Atherosclerosis basics (Pathology link): LDL oxidation in the vessel wall → macrophage engulfment → foam cell → fatty streak → fibrous plaque → rupture → acute coronary syndrome.
Pathophysiology of Dyslipidaemia: The Lipid–CVD Link
Dyslipidaemia refers to abnormal levels of lipids or lipoproteins in the blood — most clinically relevant is elevation of LDL-cholesterol, although raised triglycerides and low HDL-cholesterol independently contribute to cardiovascular risk. The pathophysiological link between dyslipidaemia and atherosclerotic cardiovascular disease (ASCVD) is well-established and causal: LDL particles penetrate the subendothelial space, where they undergo oxidative modification to oxidised LDL (ox-LDL). This ox-LDL is engulfed by macrophages that cannot down-regulate scavenger receptor activity, producing foam cells that accumulate to form the fatty streak — the earliest visible atheromatous lesion. Over years, foam cells, smooth-muscle cell proliferation, and collagen deposition form a fibrous plaque with a lipid-rich necrotic core covered by a fibrous cap. If the cap ruptures, the exposed collagen and tissue factor trigger platelet aggregation and thrombus formation, causing acute myocardial infarction or stroke. This mechanism explains why LDL reduction — regardless of the drug used — is the central therapeutic target.
Every 1 mmol/L (~39 mg/dL) reduction in LDL-C reduces the relative risk of a major cardiovascular event by approximately 22% (Cholesterol Treatment Trialists' Collaboration meta-analysis of >170,000 participants), with the benefit proportional to the absolute LDL reduction and largely independent of the agent used. Beyond LDL, hypertriglyceridaemia (particularly TG >500 mg/dL) is associated with acute pancreatitis — a more immediate indication for lipid-lowering therapy with fibrates. Low HDL-C (<40 mg/dL in men, <50 mg/dL in women) is an independent cardiovascular risk marker; raising HDL has proven harder than predicted — niacin and CETP inhibitors failed to translate their HDL-raising effect into clinical benefit.
IMPORTANT: Both primary dyslipidaemias (familial hypercholesterolaemia — heterozygous FH with LDL-R mutations; familial combined; rare lipoprotein lipase deficiency) and secondary dyslipidaemias (hypothyroidism, nephrotic syndrome, diabetes mellitus, alcohol excess, drugs) must be recognised, because the secondary cause demands treatment alongside the lipid-lowering strategy.
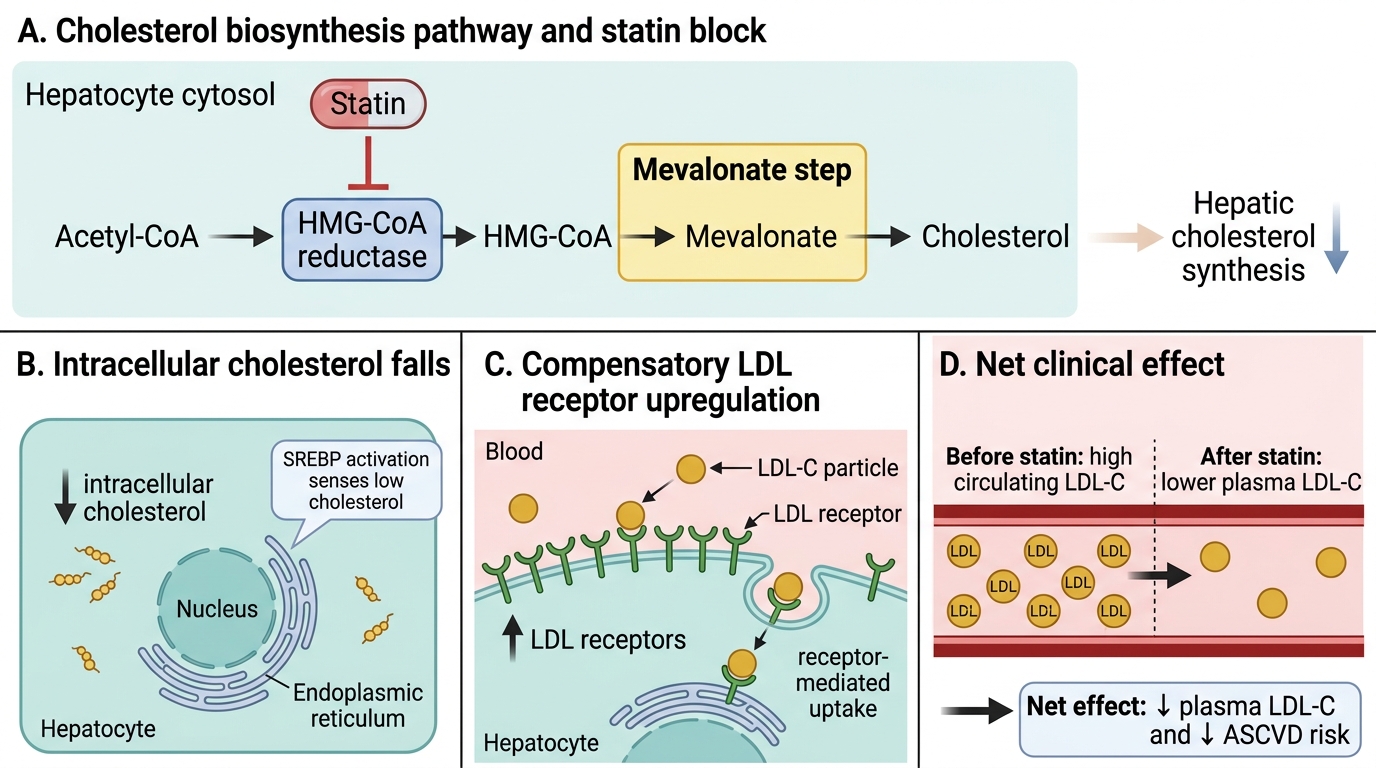
Mechanism of Statin-Induced LDL Lowering
Therapeutic Goals and Treatment Decision Framework
The treatment goal in dyslipidaemia is defined by the patient's absolute cardiovascular risk, not by the lipid value in isolation. Modern guidelines use a tiered risk framework:
Very high risk (established ASCVD — prior MI, stroke, peripheral arterial disease, or 10-year CVD risk >20%): LDL target <70 mg/dL (1.8 mmol/L); secondary prevention demands high-intensity statin therapy.
High risk (diabetes with target-organ damage, severely elevated single risk factor such as LDL >190 mg/dL or FH, or 10-year CVD risk 10–20%): LDL target <100 mg/dL (2.6 mmol/L); high-intensity statin usually needed.
Moderate risk (10-year CVD risk <10%): LDL target <116 mg/dL (3.0 mmol/L); lifestyle intervention first, moderate-intensity statin if targets not met after 3 months.
Before starting pharmacotherapy, lifestyle modification is mandatory and synergistic: a diet low in saturated fat, increased soluble fibre (psyllium, oat bran), aerobic exercise (150 min/week of moderate intensity), smoking cessation, and weight reduction. These can reduce LDL-C by 10–15% and are never replaced by drugs, only supplemented.
Triglyceride management: TG 200–500 mg/dL with established CVD — add icosapentaenoic acid (EPA, 4 g/day, REDUCE-IT trial) to statin therapy for incremental MACE reduction. TG >500 mg/dL — fibrate therapy first to prevent pancreatitis, even before statin.
The decision algorithm: start with the risk category → choose appropriate LDL target → if lifestyle insufficient, initiate statin at intensity to achieve ≥50% LDL reduction (high-intensity) or 30–49% reduction (moderate) → if target LDL not achieved, add ezetimibe → if still not at target in very-high risk, consider PCSK9 inhibitor.
Classification of Lipid-Lowering Drugs
Lipid-lowering drugs are mechanistically diverse, acting at different points in cholesterol synthesis, absorption, or excretion. The seven major classes and their primary lipid effects are:
- HMG-CoA reductase inhibitors (statins): ↓LDL 20–60%, modest ↑HDL, modest ↓TG. First-line for hypercholesterolaemia.
- Fibrates (PPAR-α agonists): ↓TG 30–50%, ↑HDL 10–20%, modest ↓LDL. First-line for severe hypertriglyceridaemia.
- Bile-acid sequestrants (BAS): ↓LDL 15–30%; may raise TG — avoid in hypertriglyceridaemia. Safe in pregnancy.
- Cholesterol-absorption inhibitors (ezetimibe): ↓LDL 15–20%. Additive with statins; used in combination when statin alone insufficient.
- PCSK9 inhibitors (monoclonal antibodies): ↓LDL 50–60%. For familial hypercholesterolaemia or intolerance to adequate statin.
- Niacin (nicotinic acid): ↑HDL 15–35%, ↓TG 20–50%, ↓LDL 15–20%. Largely superseded due to adverse effects and failure of outcome trials.
- Omega-3 fatty acids (EPA/DHA): ↓TG 20–50%. EPA (icosapentaenoic acid, 4 g/day) shown to reduce MACE in REDUCE-IT trial in statin-treated high-CV-risk patients with TG 135–499 mg/dL.
IMPORTANT: Beyond drugs that treat dyslipidaemia, PH4.11 specifically requires that you enumerate drugs that cause dyslipidaemia:
- Thiazide diuretics — raise LDL and TG (at high doses), reduce HDL; dose-dependent and partly reversible
- Non-selective β-blockers (propranolol, nadolol) — raise TG, lower HDL; cardioselective agents (atenolol, bisoprolol) have much lesser effect
- Corticosteroids — raise LDL, TG, and sometimes HDL; mechanism involves increased hepatic VLDL synthesis
- Oestrogens / oral contraceptives — raise TG (particularly with oral oestrogens); combined OCPs raise LDL slightly
- Retinoids (isotretinoin) — markedly raise TG and LDL, lower HDL; monitor lipids monthly during therapy
- Antiretrovirals — protease inhibitors (ritonavir, lopinavir) — raise TG and LDL via inhibition of VLDL clearance
- Ciclosporin — raises LDL; mechanism involves reduced hepatic LDL-R activity and reduced bile-acid synthesis
IMPORTANT: Recall that many of these dyslipidaemia-inducing drugs are necessary for their primary indication — the clinician must monitor lipids and add lipid-lowering therapy where required.
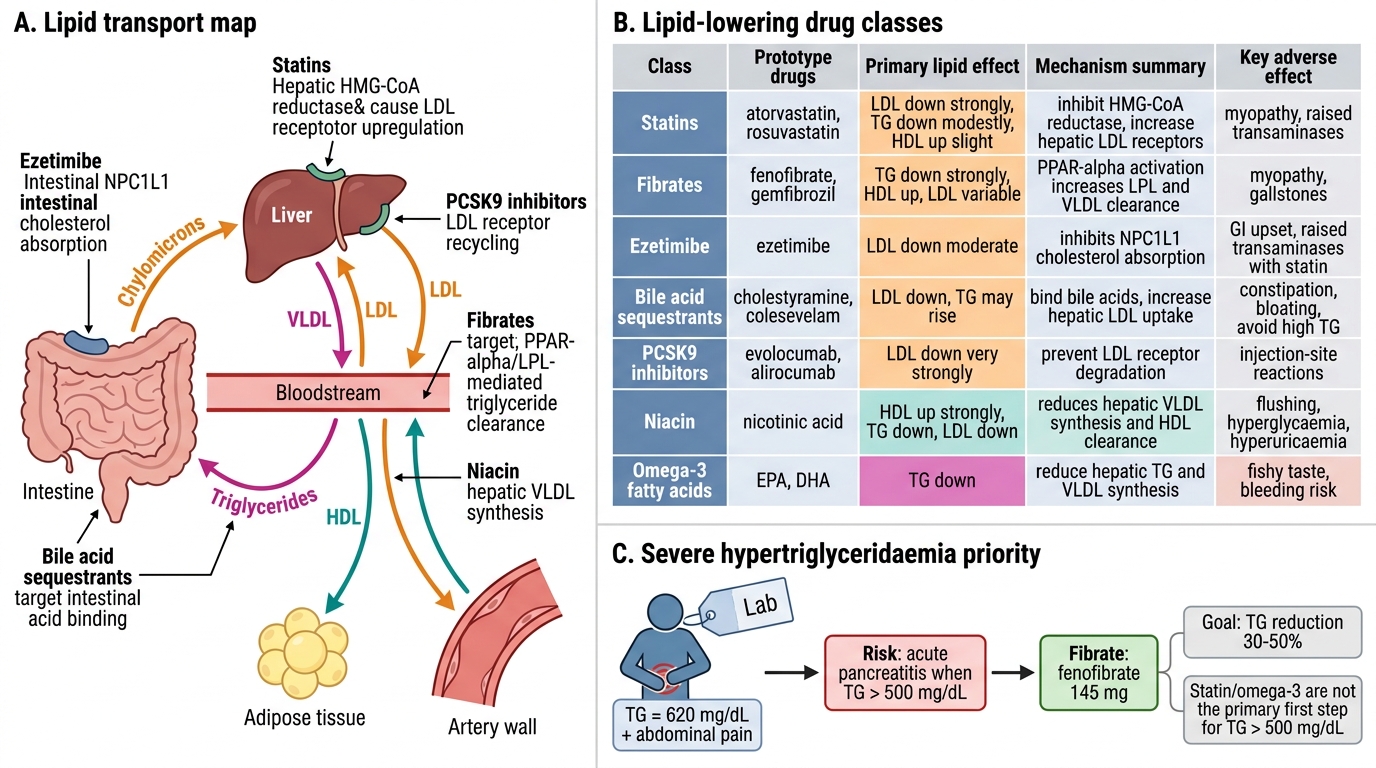
Lipid-Lowering Drug Classes and Severe Hypertriglyceridaemia
SELF-CHECK
Which drug class is MOST appropriate as first-line pharmacotherapy in a patient presenting with serum triglycerides of 620 mg/dL and abdominal pain?
A. High-intensity statin (atorvastatin 80 mg)
B. Fibrate (fenofibrate 145 mg)
C. Ezetimibe 10 mg
D. Omega-3 fatty acids 4 g/day
Reveal Answer
Answer: B. Fibrate (fenofibrate 145 mg)
When triglycerides exceed 500 mg/dL, the priority is prevention of acute pancreatitis — fibrates (PPAR-α agonists) reduce triglycerides by 30–50% and are first-line. Statins are not the primary agents for severe hypertriglyceridaemia and may actually raise TG slightly in some patients. Omega-3 fatty acids (4 g/day EPA) are appropriate for moderate hypertriglyceridaemia (135–499 mg/dL) in statin-treated patients but do not replace fibrates for TG >500 mg/dL.