Page 8 of 33
PH4.6 | PH4.6 | Renin Angiotensin Aldosterone System Modulators — SDL Guide
Learning Objectives
- Explain the physiology of the renin-angiotensin-aldosterone system (RAAS) and its role in blood pressure regulation, cardiac remodelling, and renal function.
- Classify RAAS modulators by site of action and describe the pharmacokinetics, pharmacodynamics, therapeutic uses, and adverse drug reactions of each class.
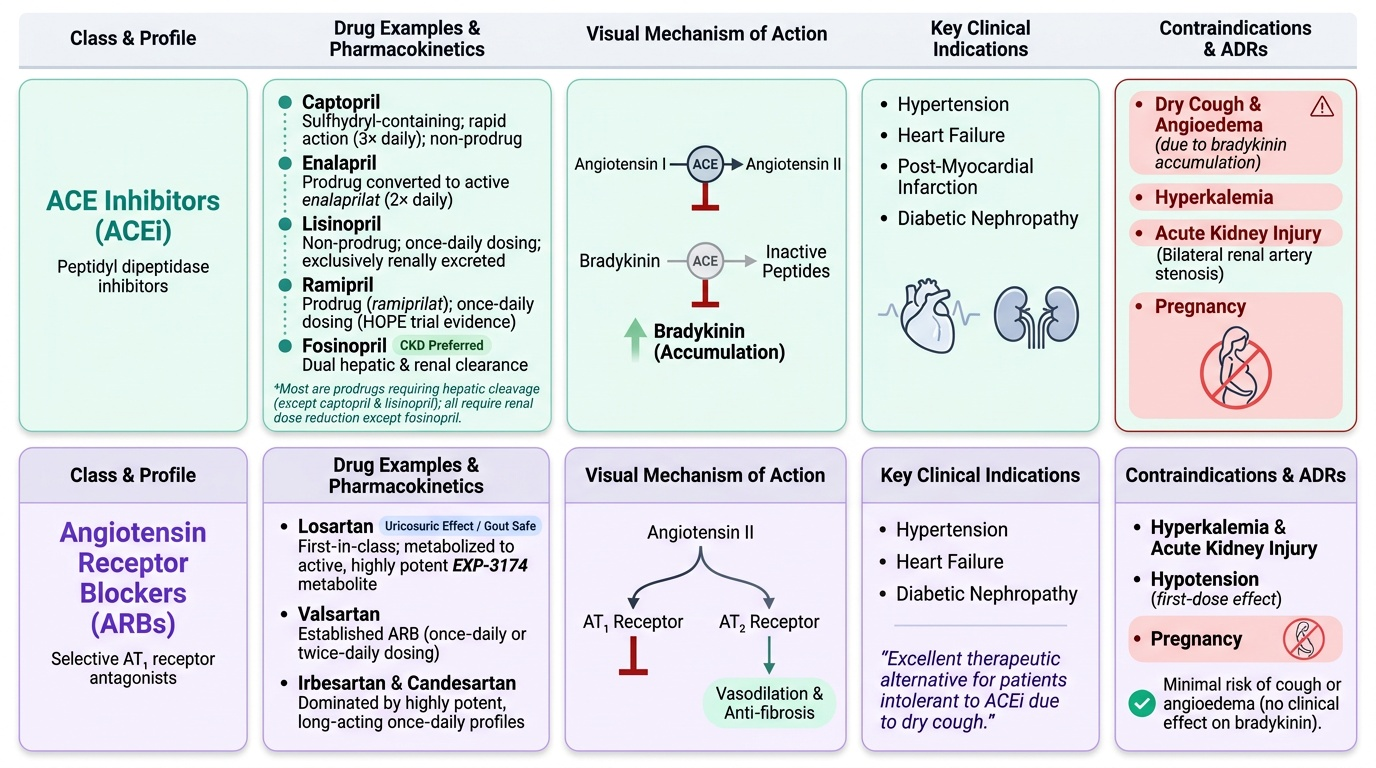
- Identify the key contraindications of ACEi and ARBs (pregnancy, bilateral renal artery stenosis) and distinguish their ADR profiles.
- Select the appropriate RAAS modulator for given clinical scenarios including heart failure, hypertension with diabetes, and CKD.
INSTRUCTIONS
The renin-angiotensin-aldosterone system is one of the most therapeutically targeted pathways in cardiovascular medicine. ACE inhibitors and ARBs are prescribed in tens of millions of patients worldwide for hypertension, heart failure, chronic kidney disease, and post-myocardial infarction remodelling. More recently, the ARNI (sacubitril/valsartan) has transformed heart failure outcomes. Understanding the full RAAS cascade — not just the drugs but their specific interaction points — allows you to reason through drug interactions, contraindications, and monitoring requirements rather than memorising disconnected facts.
References
- Tripathi KD. Essentials of Medical Pharmacology, 9th ed., Ch. 39 (Renin-Angiotensin System and Drugs) (textbook)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, 14th ed., Ch. 26 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 58-year-old man with type 2 diabetes and hypertension is found on a routine urine examination to have microalbuminuria (urine albumin:creatinine ratio 55 mg/g). His serum creatinine is 1.4 mg/dL (eGFR ~52 mL/min/1.73m²). Blood pressure is 148/88 mmHg despite lifestyle modification. He asks you: 'My father had kidney failure from his diabetes. Can we prevent that happening to me?' You prescribe ramipril 5 mg daily. Three weeks later he calls complaining of a persistent dry cough. He wants to stop the medicine. How do you explain why the cough occurs, and what is your alternative prescribing strategy to still protect his kidneys?
WHY THIS MATTERS
ACE inhibitors and ARBs are among the most prescribed drug classes in all of medicine. In India alone, tens of millions of patients with hypertension, diabetes, and heart failure take them daily. They are the only drugs with both antihypertensive and nephroprotective properties — making them uniquely positioned in diabetic nephropathy. The newer ARNI (sacubitril/valsartan) is now first-line for HFrEF after the PARADIGM-HF trial showed it outperforms ACEi. Understanding the cascade these drugs target — why ACEi cause cough and ARBs don't, why both are dangerous in pregnancy, why the ARNI requires a washout period — equips you to prescribe these agents safely and explain them convincingly to patients.
RECALL
Recall from physiology (PY) the RAAS regulatory loop:
- Renin release from juxtaglomerular (JG) cells of the afferent arteriole is triggered by: low renal perfusion pressure, low tubular sodium (sensed at macula densa), and sympathetic nervous system activation (β1 receptors on JG cells → renin release).
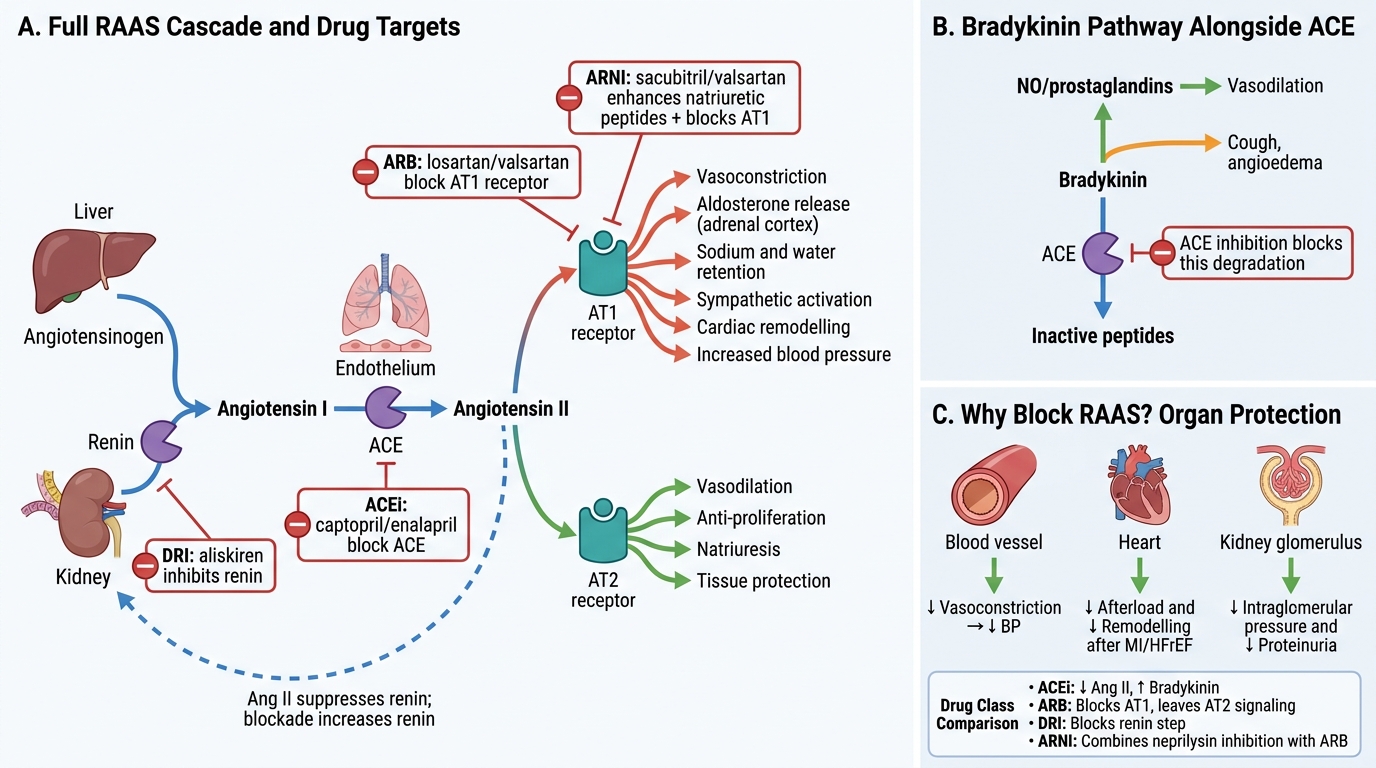
- Cascade: Renin cleaves angiotensinogen (a glycoprotein synthesised in the liver) → angiotensin I (Ang I, a decapeptide) → ACE (found mainly in pulmonary vascular endothelium) converts Ang I → angiotensin II (Ang II, an octapeptide) — the primary effector of the RAAS.
- Ang II effects via AT1 receptor: vasoconstriction (direct), aldosterone secretion (→ Na retention, K loss), sympathetic facilitation, cardiac and vascular remodelling (hypertrophy, fibrosis), renal efferent arteriolar vasoconstriction (maintains GFR but increases glomerular pressure).
- AT2 receptor (less well understood): vasodilation, anti-proliferative, anti-fibrotic — generally beneficial, opposing AT1 effects.
- Bradykinin: ACE is also a kininase — it degrades bradykinin to inactive fragments. ACEi block this → bradykinin accumulates.
RAAS Physiology: Cascade, Effectors, and Feedback
The renin-angiotensin-aldosterone system (RAAS) is a hormonal cascade that serves as the body's primary long-term regulator of blood pressure, extracellular fluid volume, and electrolyte homeostasis. Its pathological activation — chronic, inappropriately elevated RAAS activity — drives hypertension, accelerates cardiac remodelling, and mediates progressive kidney damage in diabetic nephropathy and chronic kidney disease. This makes the RAAS cascade one of the most productive therapeutic targets in medicine.
The cascade begins with renin, a protease secreted by juxtaglomerular cells of the afferent arteriole in response to reduced renal perfusion pressure, reduced sodium delivery to the macula densa, or β1-adrenoceptor stimulation. Renin cleaves the plasma glycoprotein angiotensinogen (produced by the liver, constitutively high) to produce the decapeptide angiotensin I (Ang I), which is biologically inactive. Angiotensin-converting enzyme (ACE), a carboxypeptidase concentrated on pulmonary and renal vascular endothelium, cleaves two amino acids from Ang I to produce angiotensin II (Ang II) — the primary active effector.
Ang II exerts its main cardiovascular effects through the AT1 receptor: (1) potent vasoconstriction of systemic arterioles → increased total peripheral resistance and BP; (2) stimulation of aldosterone release from the adrenal cortex → Na and water retention; (3) direct action on renal efferent arterioles (vasoconstriction) → maintained GFR despite systemic hypotension, but at the cost of increased glomerular capillary pressure → glomerular injury in the long term; (4) cardiac and vascular remodelling — myocyte hypertrophy and interstitial fibrosis contributing to ventricular dysfunction in heart failure. Ang II also acts via AT2 receptors — expressed predominantly in fetal tissue and upregulated in disease — to produce vasodilation, anti-proliferative, and anti-fibrotic effects that generally counterbalance AT1 signalling.
ACE has a second substrate: bradykinin, a vasodilatory and pain-mediating peptide. Under normal conditions, ACE rapidly degrades bradykinin. ACEi block this degradation → bradykinin accumulates → prostaglandin and nitric oxide release → vasodilation (beneficial), but also → dry cough (pulmonary bradykinin) and potentially angioedema (mast cell activation). This is the mechanistic basis for ACEi-specific ADRs.
IMPORTANT: The negative feedback loop is critical: Ang II and aldosterone suppress renin release (short-loop feedback). When ACEi or ARBs block downstream Ang II, this feedback is lost → reactive hyperreninaemia — elevated renin levels during therapy, which is pharmacologically manageable but diagnostically confusing.
RAAS Cascade, Drug Targets, and Bradykinin Pathway
Therapeutic Goals: Why Block the RAAS?
The therapeutic goals of RAAS blockade extend well beyond simple blood pressure reduction. This is the critical insight that makes ACEi and ARBs 'organ-protective' drugs rather than mere antihypertensives.
1. Blood pressure control: By reducing Ang II-mediated vasoconstriction and aldosterone-driven sodium retention, RAAS modulators lower both systolic and diastolic BP. They are effective antihypertensives across all hypertension grades and are particularly synergistic with diuretics (which activate RAAS → more to block) and calcium channel blockers.
2. Cardiac remodelling prevention (post-MI and HFrEF): After myocardial infarction, RAAS activation mediates harmful ventricular remodelling — the maladaptive changes in heart geometry and composition that lead to progressive heart failure. ACEi started within 24–48 hours of MI reduce infarct expansion, prevent pathological ventricular dilatation, and reduce mortality. In HFrEF (LVEF <40%), chronic RAAS blockade reduces afterload, reverses adverse remodelling, improves ejection fraction, and prolongs survival (CONSENSUS, SOLVD trials for ACEi; CHARM, Val-HeFT for ARBs). The ARNI sacubitril/valsartan has superseded ACEi in HFrEF based on PARADIGM-HF (20% reduction in CV death + HF hospitalisation versus enalapril).
3. Nephroprotection in diabetic nephropathy and CKD: Ang II-driven efferent arteriolar vasoconstriction increases intraglomerular pressure — the mechanism of glomerular injury in diabetic nephropathy. ACEi and ARBs reduce intraglomerular pressure beyond their systemic BP lowering effect, independently slowing the progression of microalbuminuria to macroalbuminuria and macroalbuminuria to ESRD. This nephroprotective effect is a CLASS effect of ACEi and ARBs — the drug of first choice in hypertension with diabetes or CKD with proteinuria.
4. Secondary prevention post-MI (high-risk patients): The HOPE trial (ramipril in high-CV-risk patients with normal ejection fraction) showed a 22% MACE reduction, demonstrating benefits beyond BP lowering — attributed to coronary vasodilation, endothelial function improvement, and anti-atherosclerotic plaque effects of ACEi.
Classification of RAAS Modulators
RAAS modulators are classified by the step in the cascade at which they intervene, and each class has a distinct pharmacological profile and ADR signature.
1. ACE inhibitors (ACEi): Block conversion of Ang I to Ang II AND simultaneously block bradykinin degradation. Prototypes: captopril (first-in-class, sulfhydryl group, 3× daily), enalapril (prodrug → active enalaprilat, 2× daily), lisinopril (not a prodrug, once daily, renally excreted), ramipril (prodrug → ramiprilat, once daily, evidence in HOPE trial), perindopril, quinapril, fosinopril (dual hepatic+renal elimination — safer in severe CKD). Most are prodrugs (ethyl ester promoieties converted to active diacid in liver) except captopril and lisinopril. All are cleared renally — dose reduction in CKD (fosinopril is exception).
2. Angiotensin receptor blockers (ARBs): Selectively block AT1 receptors — Ang II is still produced (no blockade of ACE) and can stimulate the unblocked AT2 receptor (vasodilation, anti-fibrosis). Prototypes: losartan (first ARB; active metabolite EXP-3174 more potent, with uricosuric effect — useful in gout), valsartan, irbesartan, candesartan, telmisartan (longest half-life, once daily, also a weak PPAR-γ agonist), olmesartan. ARBs do NOT inhibit bradykinin degradation → no cough (mechanistic advantage) → used when ACEi-induced cough is intolerable. Angioedema is rare (estimated <0.5% vs ~1-2% with ACEi) because bradykinin accumulation is not the mechanism.
3. Direct renin inhibitors (DRIs): Aliskiren inhibits renin directly at the cascade's first step → blocks the entire pathway. The ALTITUDE trial (aliskiren added to ACEi/ARB in type 2 diabetes with CKD or CVD) was stopped early due to increased renal failure, hypotension, and hyperkalaemia — aliskiren should NOT be combined with ACEi or ARBs (triple RAAS blockade). Limited clinical use.
4. Angiotensin receptor-neprilysin inhibitors (ARNIs): Sacubitril/valsartan (Entresto): sacubitril is a prodrug converted to LBQ657, a neprilysin inhibitor — neprilysin is the enzyme that degrades natriuretic peptides (ANP, BNP) and bradykinin. Inhibiting neprilysin → increases ANP and BNP → vasodilation, natriuresis, anti-fibrosis, and anti-hypertrophic effects complementing AT1 blockade by valsartan. Must NOT be combined with ACEi — bradykinin accumulates from both ACEi (blocks bradykinin degradation by ACE) and sacubitril (blocks bradykinin degradation by neprilysin) → dangerous angioedema. 36-hour washout required when switching from ACEi to sacubitril/valsartan.
IMPORTANT: The PARADIGM-HF trial showed sacubitril/valsartan reduced the composite of CV death or HF hospitalisation by 20% versus enalapril in HFrEF (LVEF ≤40%). Most international guidelines now recommend ARNI as the preferred RAAS agent in HFrEF if tolerated.
Provided image
SELF-CHECK
A 45-year-old woman with hypertension and type 2 diabetes starts enalapril 5 mg once daily. Three weeks later she develops a troublesome dry cough. The most appropriate next step is:
A. Continue enalapril — the cough will resolve within 2 weeks
B. Switch to losartan (an ARB) — same nephroprotective benefit without cough
C. Switch to amlodipine — equal nephroprotection in diabetic nephropathy
D. Add codeine linctus to suppress the cough and continue enalapril
Reveal Answer
Answer: B. Switch to losartan (an ARB) — same nephroprotective benefit without cough
ACEi-induced cough is a class effect caused by bradykinin accumulation (ACE also degrades bradykinin; when blocked, bradykinin stimulates pulmonary C-fibres → persistent dry cough). It does not resolve with time — affected patients must switch. ARBs selectively block AT1 receptors without inhibiting ACE-mediated bradykinin degradation → no bradykinin accumulation → no cough. ARBs have equivalent nephroprotective efficacy in diabetic nephropathy (IRMA-2, IDNT, RENAAL trials). Amlodipine (calcium channel blocker) is effective for hypertension but has less nephroprotective evidence in proteinuric diabetic nephropathy. Codeine is inappropriate and does not address the drug cause.