Page 17 of 30
PH10.{5,11-12} | PH10.{5,11-12} | Legal, Ethical and Regulatory Frameworks for Drug Use — SDL Guide — SDL Guide (Part 2)
Drug Development and Clinical Trials: The Path from Molecule to Medicine
Every drug currently in clinical use was once an untested molecule. Drug development is a systematic, multi-stage process that takes approximately 10–15 years and costs hundreds of millions of dollars — and the vast majority of candidate molecules fail before reaching patients. Understanding this process helps prescribers appreciate why evidence hierarchies matter and what 'approved for this indication' actually guarantees.
The drug development pipeline has two broad phases:
Pre-clinical phase:
Synthesis of candidate molecules, in vitro testing (cell culture), and in vivo animal testing for pharmacological activity, acute and chronic toxicity, teratogenicity, mutagenicity, and basic pharmacokinetics. Only ~1 in 5000–10,000 candidate molecules that enter pre-clinical testing eventually reach market approval.
Clinical trial phases:
Phase I — Safety, PK, first-in-human:
- Objective: Is the drug safe in humans? What dose range is tolerable? What are the PK parameters (Vd, t½, Cmax)?
- Population: 20–80 healthy volunteers (exception: oncology Phase I trials enrol patients with cancer, as toxicity in healthy volunteers is not acceptable)
- Primary outcome: Maximum tolerated dose, dose-limiting toxicities, safety signals
Phase II — Efficacy signal and dose-finding:
- Objective: Does the drug work in the target disease? What is the optimal dose?
- Population: 100–300 patients with the target disease
- Design: Often small RCTs or dose-ranging studies
- Primary outcome: Efficacy signal (proof of concept), dose selection for Phase III
Phase III — Confirmatory efficacy and safety:
- Objective: Is the drug superior (or non-inferior) to existing standard-of-care treatment?
- Population: 300–3,000+ patients with the target disease, representative of clinical practice
- Design: Large, double-blind, randomised controlled trials — this is the basis for marketing authorisation
- Primary outcome: Clinical endpoints (survival, hospitalisation, cure rate); safety at scale
Phase IV — Post-marketing surveillance:
- Objective: Ongoing safety monitoring after market approval; identification of rare adverse effects (1 in 10,000+ patients), long-term outcomes, real-world effectiveness in populations excluded from Phase III
- Population: All patients prescribed the drug post-approval — effectively millions
- Regulatory requirement: Pharmacovigilance reporting to CDSCO/WHO VigiBase
In India, CDSCO requires sponsors to submit a Clinical Trial Application (CTA) before beginning trials. The ethical review must be approved by an Institutional Ethics Committee (IEC) accredited by the New Drugs and Clinical Trials Rules 2019.
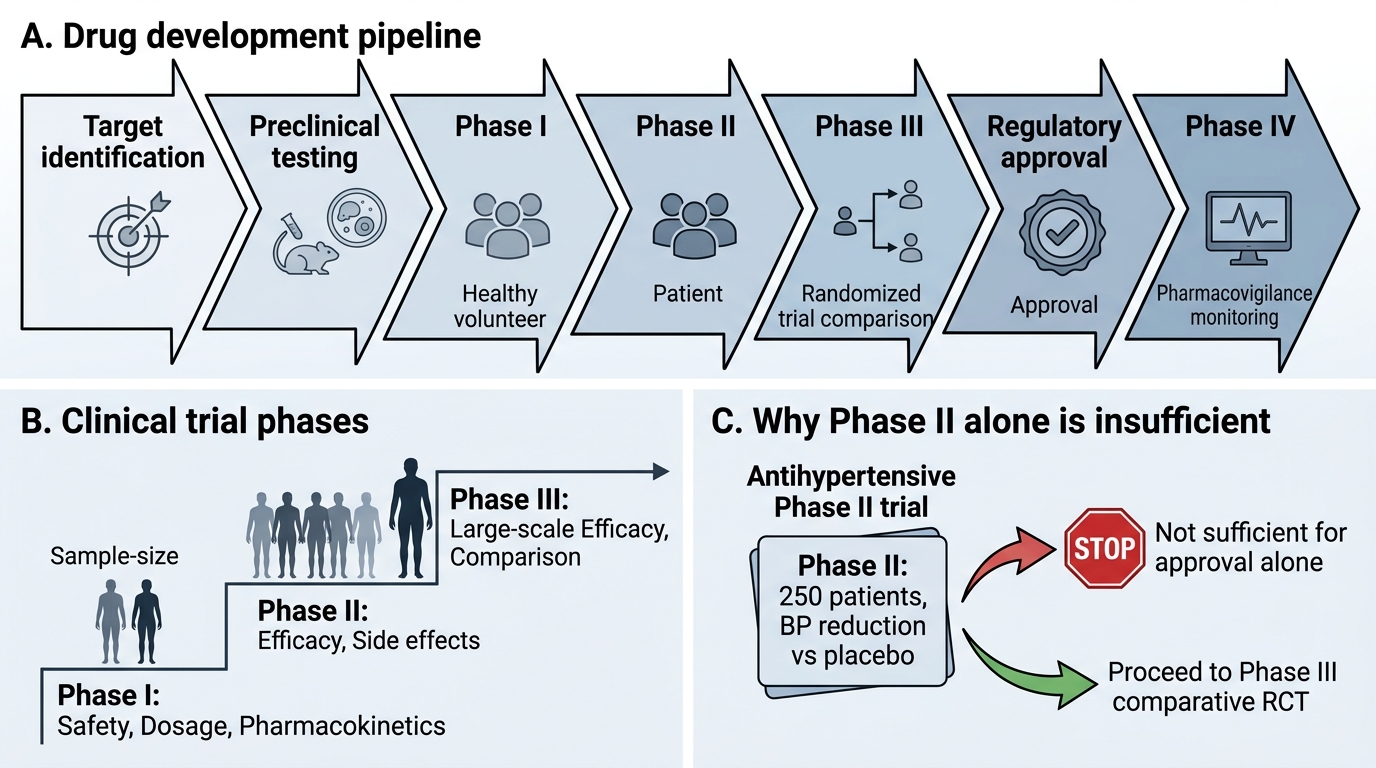
Drug Development Pipeline
SELF-CHECK
A pharmaceutical company conducts a Phase II trial of a new antihypertensive in 250 patients with hypertension. The trial shows a statistically significant blood pressure reduction compared to placebo. The company immediately applies for marketing approval based on this result. Which statement BEST explains why CDSCO would not approve based on Phase II data alone?
A. A) Phase II trials cannot be conducted in India under the Drugs and Cosmetics Act
B. B) Phase II trials use placebo comparators — CDSCO requires an active comparator study, which is a Phase II requirement
C. C) Phase II provides proof of concept and dose finding, but marketing approval requires Phase III evidence: comparative efficacy against standard-of-care at scale, and safety data from a large representative population
D. D) The company needs Phase IV data before approval — Phase III is optional for new chemical entities
Reveal Answer
Answer: C. C) Phase II provides proof of concept and dose finding, but marketing approval requires Phase III evidence: comparative efficacy against standard-of-care at scale, and safety data from a large representative population
C is correct. Phase II data establishes proof of concept and guides dose selection, but regulatory approval requires Phase III confirmatory data: large-scale RCTs demonstrating safety and efficacy against standard-of-care treatment in a representative patient population. Phase II is typically underpowered to detect uncommon adverse effects and does not provide the comparative evidence needed to establish where the new drug fits in the treatment armamentarium. CDSCO's New Drugs and Clinical Trials Rules 2019 require complete Phase III data as part of the NDA (New Drug Application) submission.
The Role of Research in Developing New Drugs: Ethical Considerations
Clinical research is the mechanism by which pharmacotherapy advances — every improvement in cancer survival, HIV treatment, and cardiovascular prevention rests on decades of clinical trials. But research involving human participants carries inherent ethical risks, and the history of medicine includes grave abuses that have shaped the international ethical framework.
The cornerstone of research ethics is the Declaration of Helsinki (World Medical Association, 1964, last revised 2013): a statement of ethical principles for medical research involving human subjects. Its key principles are:
1. The interests of the individual research participant must always take precedence over the interests of science and society
2. Informed consent is required from all research participants — they must understand the purpose, risks, and benefits of participation and must be free to refuse or withdraw at any time without penalty
3. Independent ethics review by an Institutional Review Board (IRB) or Institutional Ethics Committee (IEC) is mandatory before any clinical research begins
4. Research must be conducted only by qualified persons — principal investigators are responsible for the welfare of participants
5. Good Clinical Practice (GCP) — the ICH E6(R2) guideline defines the international quality standard for designing, conducting, recording, and reporting clinical trials involving human subjects; it ensures data integrity and participant protection simultaneously
Indian regulatory framework for research:
The ICMR National Ethical Guidelines for Biomedical and Health Research Involving Human Participants (2017) is the foundational document for research ethics in India. It addresses: consent processes (including surrogate consent for incapacitated subjects), research with vulnerable populations (children, prisoners, pregnant women, economically disadvantaged), payment for participation, data and sample management, and post-trial obligations.
The New Drugs and Clinical Trials Rules 2019 (CDSCO) replaced the earlier Schedule Y provisions and introduced: accelerated approval pathways for rare diseases and unmet needs; requirements for local Phase I data for new molecules; compensation for trial-related injuries; and requirements for trial registration with CTRI (Clinical Trials Registry India).
The doctor's role in research:
As a future clinician, you may be invited to enrol your patients in clinical trials. Key ethical obligations:
- Do not enrol patients without truly independent informed consent — research participation must not be conflated with clinical care
- Conflict of interest: payment from sponsors for each enrolled patient creates an incentive that must be disclosed and managed
- Publication ethics: report all outcomes including negative results — selective reporting (publishing only positive trials) is a major driver of publication bias and distorts the evidence base
- Research misconduct (fabrication, falsification, plagiarism) carries criminal penalties in India and permanent loss of medical registration
Evaluating the Regulatory and Ethical Dimensions of Drug Choices
Applying the regulatory and ethical frameworks to real prescribing decisions requires integrating legal knowledge with clinical judgment. Three complex scenarios illustrate the kinds of decisions that arise in clinical practice.
Scenario 1 — Off-label prescribing in pregnancy:
A 30-year-old pregnant woman (16 weeks) with hyperemesis gravidarum has not responded to approved first-line treatments (ginger, vitamin B6, promethazine). A gastroenterologist suggests ondansetron (a 5-HT3 antagonist approved for chemotherapy-induced nausea, not approved specifically for hyperemesis in pregnancy in India). Ondansetron has some evidence from small RCTs for hyperemesis. What do you do?
Legal status: off-label prescribing is permitted; ondansetron is not contraindicated but has limited pregnancy safety data (some studies showed a minor signal for cardiac defects in first trimester — risk is debated but risk-benefit may favour it in severe hyperemesis). Ethical obligations: informed consent documenting the off-label status, the evidence, the uncertainty, and that first-line treatments have failed; document clinical reasoning. Prescribing requires a genuine evidence base (published literature), not just tradition.
Scenario 2 — Pharmacovigilance obligation:
You prescribe a newly approved antibiotic. Two weeks later, the patient returns with unexplained hepatitis with no other cause identified. This appears to be a drug adverse reaction. What are your regulatory obligations?
All healthcare professionals have a legal and professional duty to report suspected adverse drug reactions to the CDSCO's National Pharmacovigilance Programme via the online ADR reporting form. You are also expected to report to the hospital's drug safety committee and document in the patient's record. Failure to report is not a criminal offence in India (unlike in some jurisdictions), but it is a professional obligation and contributes to the regulatory database that protects future patients.
Scenario 3 — Industry-sponsored research:
A pharmaceutical company approaches you to enrol patients in a Phase III trial of a new antibiotic. They offer ₹5,000 per enrolled patient. Before agreeing, what do you need to confirm?
(1) IEC approval for the trial at your institution; (2) CTRI registration; (3) review the trial protocol yourself — you must be satisfied the trial is scientifically and ethically sound; (4) disclose the payment arrangement to patients and your institution; (5) ensure the payment structure does not compromise patient consent — per ICMR guidelines, financial incentives to investigators must not be so large as to distort clinical judgment or consent; (6) confirm adequate injury compensation arrangements for participants.
CLINICAL PEARL
Off-label prescribing requires informed consent documentation. 'Off-label' does not mean illegal or experimental — it simply means the drug is being used outside the exact indication approved by the regulatory authority. Many established practices (e.g. aspirin for thromboprophylaxis in various contexts, metformin in PCOS) involve off-label use. The ethical and legal requirement is that the patient understands: (1) the drug is being used in a way not specifically approved for their condition; (2) the evidence base supporting the use; (3) the uncertainty and monitoring plan. Documenting this conversation in the medical record is both good practice and legal protection.
Self-Assessment: Drug Regulation, Ethics, and Drug Development
Consolidate your regulatory and ethical knowledge through the following structured exercises.
Exercise 1 — Schedule classification:
Classify each of the following drugs by their Schedule under the Drugs and Cosmetics Act and state one key additional prescription requirement (beyond basic Rx):
(a) Clonazepam (b) Amoxicillin (c) Cefoperazone-sulbactam combination
Expected answers:
(a) Clonazepam — Schedule X (benzodiazepine under NDPS Act); requirement: prescription in duplicate, quantity in words, patient name and address, prescriber registration number; pharmacist retains copy for 2 years.
(b) Amoxicillin — Schedule H; basic Rx required, pharmacist retains copy for 3 years; no additional special requirement beyond standard Schedule H.
(c) Cefoperazone-sulbactam — Schedule H1 (extended-spectrum cephalosporin combination antimicrobial); prescription in duplicate, prescriber registration required, pharmacist retains copy for 3 years; stricter documentation under H1 rules to limit inappropriate antimicrobial use.
Exercise 2 — Ethics of self-prescribing:
A colleague who is also a physician asks you to prescribe diazepam for him without examination because he is 'too busy to see his own doctor.' List three reasons why this request is professionally problematic and state what you would recommend instead.
Expected answers:
(1) No clinical examination has been performed — prescribing without examination is ethically and legally problematic; the prescriber cannot document a valid clinical indication without assessing the patient. (2) Diazepam is a Schedule X drug — prescribing requires additional documentation and a therapeutic rationale; prescribing to a colleague who is requesting a controlled drug without assessment creates potential for facilitating dependence or misuse. (3) Bias and objectivity: prescribing for a colleague creates social pressure that compromises independent clinical judgment — the NMC code advises against prescribing for close contacts except in genuine emergencies. Recommendation: the colleague should register with a general practitioner and seek appropriate medical assessment.