Page 5 of 15
PH2.7 | PH2.7 | Non-Opioid Analgesics and NSAIDs — SDL Guide — SDL Guide (Part 2)
Pharmacokinetics, Mechanism of Action, and Pharmacodynamics
Understanding the pharmacokinetic profiles of analgesics explains their dosing schedules, accumulation risks, and the rationale for choosing one drug over another for a specific clinical scenario.
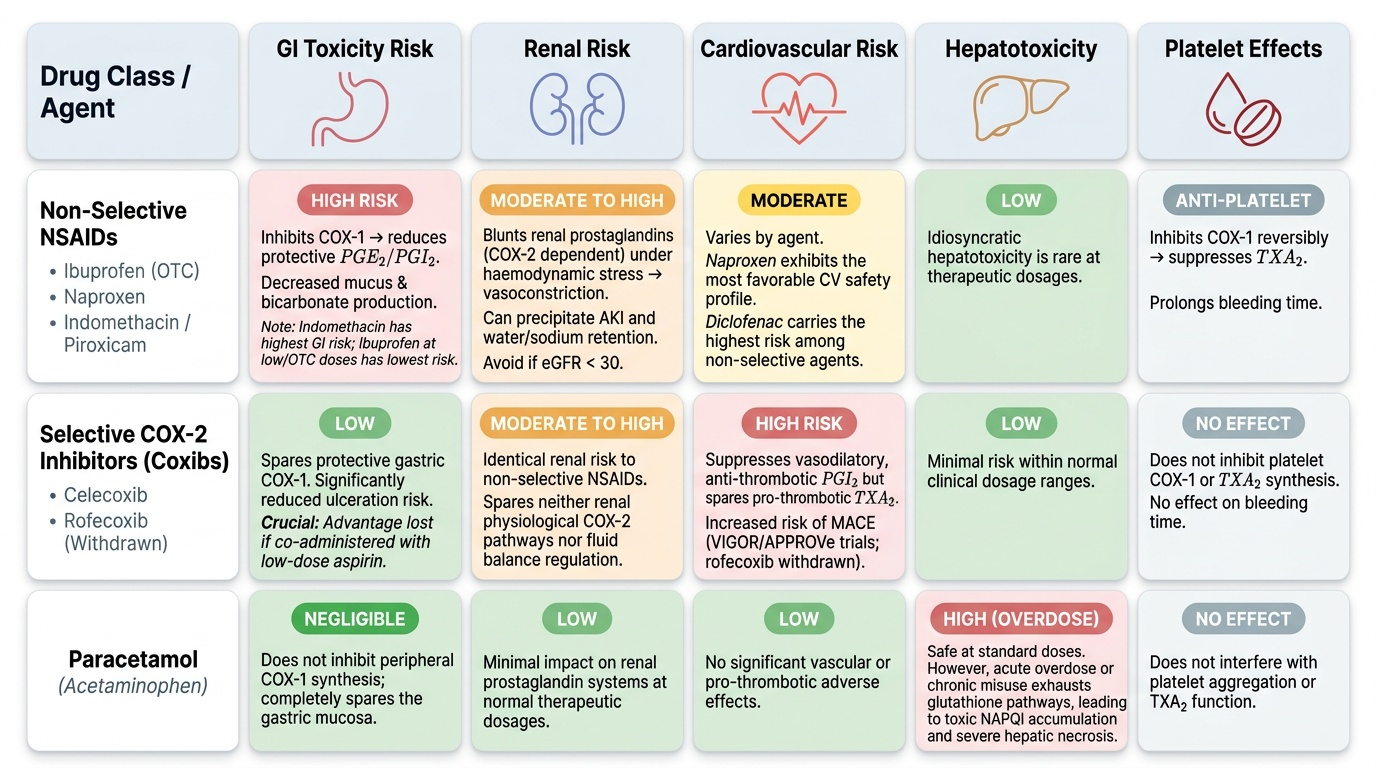
All oral NSAIDs are rapidly and completely absorbed from the upper GI tract (bioavailability typically 80–100%). They are highly protein-bound (>95% for most) — important for drug interactions with warfarin and methotrexate (displacement from albumin). They concentrate in inflamed tissue due to local pH changes (ionisation trapping). Most NSAIDs undergo hepatic metabolism (glucuronidation, oxidation) and renal excretion. Clinically important PK differences:
- Aspirin: short plasma half-life (~15–20 min) but its acetylation of platelet COX-1 is irreversible — platelets cannot regenerate COX because they have no nucleus, so the antiplatelet effect lasts the full platelet lifespan (7–10 days). This is the mechanistic basis for aspirin's irreversible platelet inhibition and why it is stopped 7–10 days before elective surgery.
- Piroxicam: long half-life (45–50 hours); once-daily dosing but accumulation risk in elderly.
- Ibuprofen: short half-life (2 hours); requires multiple daily dosing; GI-safer than indomethacin.
- Ketorolac: IM/IV/oral; powerful analgesic; maximum 5 days systemic use (risks severe GI and renal toxicity with prolonged use).
- Celecoxib: moderate half-life (11 hours); oral; metabolised by CYP2C9 (interactions with fluconazole, fluvastatin).
The mechanism of COX inhibition distinguishes aspirin from all other NSAIDs. Aspirin acetylates a serine residue (Ser530 on COX-1, Ser516 on COX-2) within the active site, permanently blocking substrate access — an irreversible covalent mechanism. All other NSAIDs are reversible competitive inhibitors, binding non-covalently and having their effect dissipate as plasma drug concentrations fall.
Selective vs non-selective inhibition: Non-selective NSAIDs inhibit both COX-1 (gastric protection, platelet TXA2) and COX-2 (inflammatory PGE2). Selective coxibs inhibit COX-2 with >50-fold greater affinity, sparing COX-1-mediated cytoprotection. However, COX-2 in the endothelium produces prostacyclin (PGI2), which inhibits platelet aggregation and promotes vasodilation. Coxibs suppress endothelial PGI2 without suppressing platelet TXA2 (COX-1-dependent), shifting the haemostatic balance toward thrombosis — the mechanistic basis of their cardiovascular risk.
Adverse Drug Reactions and Drug Interactions of NSAIDs
The adverse effects of NSAIDs follow directly from their mechanism of action and can be predicted from their COX selectivity profile. The three major organ systems at risk are the GI tract, the kidneys, and the cardiovascular system.
Gastrointestinal toxicity is the most common and clinically significant class effect of non-selective NSAIDs. Inhibition of COX-1 in the gastric mucosa reduces PGE2 and PGI2, leading to decreased mucus secretion, decreased bicarbonate production, reduced mucosal blood flow, and increased susceptibility to acid-peptic damage. This results in superficial erosions, gastric ulcers, and potentially life-threatening GI bleeding. The risk is highest with indomethacin and piroxicam; lowest with ibuprofen at OTC doses. Risk factors amplifying NSAID GI toxicity: age >65, prior peptic ulcer history, concomitant corticosteroids or anticoagulants, H. pylori infection. Coxibs significantly reduce GI ulceration risk vs non-selective NSAIDs (as shown in the CLASS trial), but this advantage is lost when low-dose aspirin is co-administered.
Renal toxicity: Prostaglandins (COX-2-derived) are critical for maintaining renal blood flow under conditions of haemodynamic stress (dehydration, heart failure, cirrhosis, hypovolaemia). NSAIDs that inhibit renal prostaglandin synthesis in these patients precipitate acute kidney injury (AKI) by inducing renal vasoconstriction. They also cause sodium and water retention, aggravating hypertension and heart failure. Contraindicated when eGFR <30 mL/min/1.73m². They also blunt the effect of thiazide and loop diuretics and increase lithium toxicity (reduce renal lithium clearance).
Cardiovascular risk: All NSAIDs (non-selective and coxibs) carry some cardiovascular risk via different mechanisms. Coxibs most prominently (PGI2 suppression without TXA2 suppression). The VIGOR trial showed a 4-fold increase in MACE with rofecoxib vs naproxen; the APPROVe trial confirmed a doubling of cardiovascular events with rofecoxib vs placebo, leading to its global withdrawal in 2004. Diclofenac has the highest cardiovascular risk among the non-selective NSAIDs per network meta-analyses. Naproxen has the most favourable cardiovascular profile among non-selective NSAIDs.
Paracetamol hepatotoxicity: Paracetamol is safe at therapeutic doses because it is primarily glucuronidated and sulfated. A small fraction is oxidised by CYP2E1 and CYP3A4 to the reactive metabolite NAPQI (N-acetyl-p-benzoquinone imine), which is rapidly conjugated with glutathione to form a non-toxic compound. In overdose (>150 mg/kg or >7.5–10 g acutely in adults), glutathione stores are exhausted, allowing NAPQI to accumulate and covalently bind hepatocyte proteins, causing centrilobular hepatic necrosis. Antidote: N-acetylcysteine (NAC), which replenishes glutathione precursors; most effective within 8 hours of ingestion.
Provided image
Key drug interactions:
- All NSAIDs + anticoagulants (warfarin): enhanced bleeding risk via platelet inhibition + protein-binding displacement
- NSAIDs + antihypertensives (especially ACE inhibitors + diuretics): reduced antihypertensive efficacy; increased AKI risk (the "triple whammy" — NSAID + ACE inhibitor + diuretic)
- Aspirin + methotrexate: displaces methotrexate from protein binding → methotrexate toxicity
- NSAIDs + lithium: reduced renal lithium clearance → lithium toxicity
- Ibuprofen + low-dose aspirin: competitive COX-1 inhibition — ibuprofen may interfere with aspirin's antiplatelet effect if taken together (take aspirin 30 min before ibuprofen, or use a non-COX-1-binding NSAID)
NSAID-exacerbated respiratory disease (aspirin-sensitive asthma): Approximately 10–20% of adult asthmatics develop bronchoconstriction with aspirin or other NSAIDs. The mechanism is COX inhibition shunting arachidonic acid into the LOX pathway, increasing leukotriene synthesis. Coxibs are generally safer in these patients (preserve COX-1 LOX-shunting effect of the standard NSAIDs is COX-2-independent, and coxibs do not block COX-1 significantly).
Aspirin in children — Reye's syndrome: Aspirin must NOT be used in children under 12 (or 16 in some guidelines) with viral illness. The combination of aspirin and viral infection (particularly varicella or influenza B) is associated with Reye's syndrome (acute encephalopathy + hepatic failure). Paracetamol is the safe antipyretic in children.
SELF-CHECK
A 68-year-old woman with osteoarthritis requires long-term NSAID therapy. She has no prior GI disease but is on low-dose aspirin for secondary cardiovascular prevention. Which strategy BEST balances her analgesia and GI safety?
A. Celecoxib alone — its COX-2 selectivity fully protects the GI tract even with aspirin co-administration
B. Indomethacin — the most potent COX inhibitor gives the best anti-inflammatory effect
C. A non-selective NSAID (e.g. naproxen) + a proton pump inhibitor, given that co-aspirin negates the GI advantage of coxibs
D. Paracetamol — it is an effective anti-inflammatory for osteoarthritis
Reveal Answer
Answer: C. A non-selective NSAID (e.g. naproxen) + a proton pump inhibitor, given that co-aspirin negates the GI advantage of coxibs
The CLASS trial demonstrated that celecoxib's GI advantage over non-selective NSAIDs is largely lost when low-dose aspirin is co-administered, because aspirin itself inhibits COX-1 in the gastric mucosa. The preferred strategy is a non-selective NSAID (naproxen is preferred for its favourable cardiovascular profile) combined with a proton pump inhibitor for gastroprotection. Indomethacin has a high ADR profile and is not a first-choice long-term NSAID. Paracetamol lacks meaningful anti-inflammatory activity and is insufficient for osteoarthritis with active synovitis.
Therapeutic Uses, Contraindications, and Clinical Decision-Making
Rational prescribing of non-opioid analgesics requires matching the drug to the clinical scenario, taking into account pain type, severity, co-morbidities, and risk factors. The WHO analgesic ladder places non-opioid analgesics at Step 1, but the choice within this step requires pharmacological reasoning.
Paracetamol first: For mild-to-moderate pain without inflammation — headache, post-procedure pain, mild musculoskeletal ache, fever — paracetamol (500–1000 mg every 4–6 hours, maximum 4 g/day) is the safest first choice. It is free of GI, renal, and cardiovascular risks at therapeutic doses and is appropriate for the elderly, patients with renal impairment (dose-reduce), and those on anticoagulants.
Non-selective NSAID when anti-inflammation is needed: Osteoarthritis with active synovitis, acute gout, dysmenorrhoea, post-operative pain, musculoskeletal inflammation, ankylosing spondylitis — ibuprofen, naproxen, or diclofenac are appropriate. Always use the lowest effective dose for the shortest necessary duration. Co-prescribe a proton pump inhibitor (PPI) (omeprazole, pantoprazole) in patients with GI risk factors (age >65, prior ulcer, concurrent anticoagulant or corticosteroid use).
Coxib when GI risk is high and CV risk is acceptable: Celecoxib or etoricoxib in patients with high GI risk but no significant cardiovascular risk or aspirin co-medication. Do NOT prescribe coxibs to patients with established IHD, cerebrovascular disease, or peripheral arterial disease without careful benefit-risk discussion.
Aspirin — unique dual role: Low-dose aspirin (75–150 mg/day) is a antiplatelet agent, not primarily an analgesic, exploiting its irreversible COX-1 inhibition in the anucleate platelet. At high anti-inflammatory doses, aspirin causes significant GI toxicity and is no longer preferred as an anti-inflammatory agent (better NSAIDs available). Children must NOT receive aspirin (Reye's syndrome risk).
Indomethacin — reserved indications: Patent ductus arteriosus closure (inhibits PG-dependent ductal patency), gout (acute flares — though colchicine or celecoxib preferred now), ankylosing spondylitis. Its high ADR rate (GI, CNS — headache, dizziness, confusional states) limits routine use.
Ketorolac: Parenteral analgesic for moderate-to-severe acute pain (post-operative, renal colic) where an opioid-sparing effect is desired. Maximum 5 days systemic use — longer use is contraindicated due to GI and renal toxicity.
Contraindications for NSAIDs:
- Active or recent peptic ulcer disease
- Chronic kidney disease (eGFR <30 mL/min/1.73m²)
- Decompensated heart failure
- Third trimester of pregnancy (risk of premature closure of ductus arteriosus via PG inhibition; labour inhibition)
- Aspirin-sensitive asthma (NSAID-exacerbated respiratory disease)
- Aspirin in children <12 years (Reye's syndrome)
- Coxibs in established cardiovascular disease (coronary artery disease, CVA, peripheral arterial disease)
CLINICAL PEARL
The 'Triple Whammy' Combination: Prescribing an NSAID to a patient already on an ACE inhibitor (or ARB) and a diuretic creates the 'triple whammy' — three simultaneous hits on renal haemodynamics. The ACE inhibitor reduces angiotensin II-mediated efferent arteriole vasoconstriction, lowering GFR. The diuretic reduces intravascular volume, activating the renin-angiotensin-prostaglandin system to maintain GFR. The NSAID then removes the prostaglandin-mediated afferent arteriole vasodilation that was the kidneys' last compensatory mechanism — precipitating acute kidney injury. This combination is particularly dangerous in elderly, dehydrated, or heart failure patients and is a leading cause of preventable drug-induced AKI in hospital settings.
SELF-CHECK
A patient takes paracetamol 1 g four times daily regularly for chronic pain. He is also a chronic alcohol user. Why is he at increased risk of paracetamol hepatotoxicity?
A. Alcohol directly inhibits glucuronidation of paracetamol, causing accumulation
B. Alcohol induces CYP2E1, increasing NAPQI production, while also depleting hepatic glutathione stores
C. Alcohol increases COX-2 expression in the liver, enhancing paracetamol's hepatic effects
D. Alcohol reduces renal clearance of paracetamol, causing accumulation
Reveal Answer
Answer: B. Alcohol induces CYP2E1, increasing NAPQI production, while also depleting hepatic glutathione stores
Chronic alcohol use induces CYP2E1, the enzyme primarily responsible for converting paracetamol to the toxic metabolite NAPQI. Simultaneously, chronic alcohol consumption depletes hepatic glutathione stores (the defence against NAPQI). This dual hit — more NAPQI formed + less glutathione to neutralise it — means paracetamol hepatotoxicity can occur at doses considered safe in non-drinkers (doses as low as 2–4 g/day in chronic alcoholics). This is a critical prescribing consideration in patients with alcohol use disorder.