Page 36 of 46
PH8.11 | PH8.11 | Anticancer Drugs and Toxicity Amelioration — SDL Guide — SDL Guide
Learning Objectives
- Describe the types, kinetics, dynamics, adverse effects, and indications of anti-cancer drugs
- Classify anticancer drugs as cell-cycle specific (CCS) or cell-cycle non-specific (CCNS) with mechanistic justification
- Devise a plan for amelioration of key anticancer drug-induced toxicities: haemorrhagic cystitis, cardiotoxicity, methotrexate toxicity, nephrotoxicity, chemotherapy-induced nausea/vomiting, and myelosuppression
INSTRUCTIONS
Cancer kills approximately 800,000 people annually in India, with incidence rising from changing lifestyle, ageing population, and improving detection rates. Anticancer pharmacology is distinct because the drugs target rapidly dividing cells — a strategy that succeeds against tumour cells but inevitably damages normal rapidly dividing tissues (bone marrow, GI epithelium, hair follicles). This creates the therapeutic challenge: maximising tumour kill while protecting normal tissues. PH8.11 explicitly adds 'devise plan for amelioration of anticancer drug-induced toxicity' — this toxicity management component distinguishes this SDL from a simple drug list.
References
- Tripathi KD. Essentials of Medical Pharmacology, 8th ed., Ch 66 (textbook)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, 13th ed., Ch 71 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 35-year-old woman with newly diagnosed breast cancer is scheduled for AC chemotherapy (doxorubicin + cyclophosphamide). She is terrified by what she has read online: hair loss, nausea, infections, heart damage. She asks the oncologist: 'Will this treatment damage my heart permanently? I've heard doxorubicin destroys your heart.' The oncologist must explain: doxorubicin does cause cumulative dose-dependent cardiomyopathy — the lifetime cumulative dose of >450–550 mg/m² markedly increases risk. However, for her planned 4-cycle AC regimen, she will receive approximately 240 mg/m² — well within safe limits. More importantly, there are drugs (dexrazoxane) and strategies (echocardiographic monitoring) that protect the heart when higher cumulative doses are needed. And cyclophosphamide, feared for bladder damage, is administered with MESNA and aggressive hydration to prevent haemorrhagic cystitis. Pharmacological knowledge of toxicity — and its amelioration — makes the difference between a patient who stops treatment in fear and one who completes curative chemotherapy.
WHY THIS MATTERS
Anticancer drug toxicity causes patient suffering, dose reductions, and treatment discontinuation — all of which compromise outcomes. As a physician, you will manage cancer patients in every specialty: the medical officer referring a patient for chemotherapy must know its toxicity profile to monitor appropriately; the anaesthesiologist must know that bleomycin-exposed lungs are sensitive to high oxygen concentrations (hyperoxia precipitates pulmonary toxicity); the internist managing a patient during chemotherapy must recognise tumour lysis syndrome, febrile neutropenia, and anthracycline cardiomyopathy. Understanding toxicity amelioration is not an oncology subspecialty — it is core general medicine.
RECALL
From cell biology (Year 1): The cell cycle has four phases: G1 (gap 1, preparation for DNA synthesis), S (DNA synthesis), G2 (gap 2, preparation for mitosis), and M (mitosis). Quiescent cells are in G0 (resting phase). Rapidly dividing normal tissues (bone marrow, GI mucosa, hair follicles, gonads) share the vulnerability of tumour cells to drugs targeting cell division — this is the pharmacological basis for the shared toxicity. From pharmacology: alkylation is the formation of covalent bonds between a reactive alkyl group and cellular nucleophiles (DNA bases) — the mechanism of many chemotherapy agents and some chemical warfare agents (mustard gas, the prototype). Antimetabolites mimic normal cellular building blocks but cannot be used productively → cell death.
Cancer Pathobiology: Why Targeting Rapidly Dividing Cells Is the Core Strategy
Cancer is fundamentally a disease of disordered cell division — cells that bypass normal regulatory checkpoints and divide uncontrollably. The pharmacological strategy of traditional cytotoxic chemotherapy exploits a single fundamental difference between most cancer cells and most non-dividing normal cells: cancer cells divide more frequently and are therefore more often in the cell cycle than normal cells in protected tissues (neurons, hepatocytes, cardiomyocytes, which rarely divide).
The cell cycle phases and their pharmacological relevance:
- G0 (quiescent/resting): Cells not actively dividing; may re-enter the cell cycle under appropriate signals. Cancer stem cells and some treatment-resistant cells hide in G0. CCNS drugs (alkylating agents) can kill G0 cells; CCS drugs cannot.
- G1 (gap 1): Cell grows and prepares for DNA synthesis; protein synthesis active. Duration highly variable (hours to days). Some regulatory checkpoints (Rb, cyclin D/CDK4). Drugs: few specific G1-phase agents (some hormonal agents and targeted therapies).
- S phase (DNA synthesis): DNA replication occurs. Highly vulnerable to antimetabolites (methotrexate, 5-FU, cytarabine, gemcitabine) — these masquerade as nucleotide precursors and disrupt DNA replication.
- G2 (gap 2): Cell verifies DNA integrity before mitosis; DNA repair enzymes active. Bleomycin and etoposide are active here (DNA strand breaks, topoisomerase II inhibition).
- M phase (mitosis): Chromosomes separate; spindle apparatus operates. Highly vulnerable to antimitotic drugs: vinca alkaloids (prevent spindle formation by blocking tubulin polymerisation) and taxanes (prevent spindle disassembly by hyperstabilising microtubules — opposite mechanism to vincas).
Normal rapidly-dividing tissues at risk from chemotherapy:
- Bone marrow: Haematopoietic stem cells divide frequently → myelosuppression (neutropenia → infection; thrombocytopenia → bleeding; anaemia)
- GI mucosa: Enterocytes replaced every 3–5 days → mucositis (oral ulcers), diarrhoea, nausea
- Hair follicles: Keratinocytes rapidly dividing → alopecia (reversible)
- Gonads: Spermatogonia and developing follicles → infertility (discuss sperm banking before alkylating agent chemotherapy)
- Fetal tissues: Rapidly dividing → severe teratogenicity (most cytotoxic drugs contraindicated in pregnancy — especially first trimester)
Therapeutic Goals: Tumour Kill, Resistance Prevention, and Toxicity Amelioration
The therapeutic strategy of cytotoxic chemotherapy is guided by principles that mirror antimicrobial pharmacology — because cancer cells, like bacteria, develop resistance, and multiple drugs targeting different mechanisms are required for durable benefit.
Log-kill hypothesis: Each chemotherapy cycle kills a constant fraction (not a constant number) of tumour cells. If a drug kills 99% of cells per cycle, then: 10⁹ cells → 10⁷ → 10⁵ → 10³ → 10 → 0 (requires approximately 5 cycles, even if each cycle eliminates 2 logs). This explains why multiple cycles are needed and why stopping treatment early — even when the patient feels well — allows residual cancer cells to proliferate.
Combination chemotherapy rationale:
1. Different mechanisms: Combining drugs with different molecular targets (alkylating agent + antimetabolite + vinca) kills cells via multiple pathways, reducing the chance of survival for any single cell.
2. Non-overlapping toxicities: Selecting drugs whose dose-limiting toxicities affect different organs allows each drug to be given at near-maximum dose (e.g. vincristine is not myelosuppressive — it can be combined with myelosuppressive agents without worsening bone marrow toxicity).
3. CCS + CCNS together: CCNS drugs kill G0 cells (driving them into the cycle); CCS drugs then kill proliferating cells emerging from G0.
4. Resistance prevention: Multiple drugs prevent the selection of resistant clones by any single mechanism.
PH8.11 — Toxicity Amelioration as a therapeutic goal: Uniquely among the antimicrobial competencies, PH8.11 explicitly requires devising a plan for amelioration of anticancer drug toxicities. This is not a footnote — it is clinically as important as the anticancer drug itself. A patient who develops haemorrhagic cystitis from cyclophosphamide may refuse further treatment; a patient whose doxorubicin-induced cardiomyopathy goes unmonitored may develop irreversible heart failure. The amelioration plan is part of the prescription.
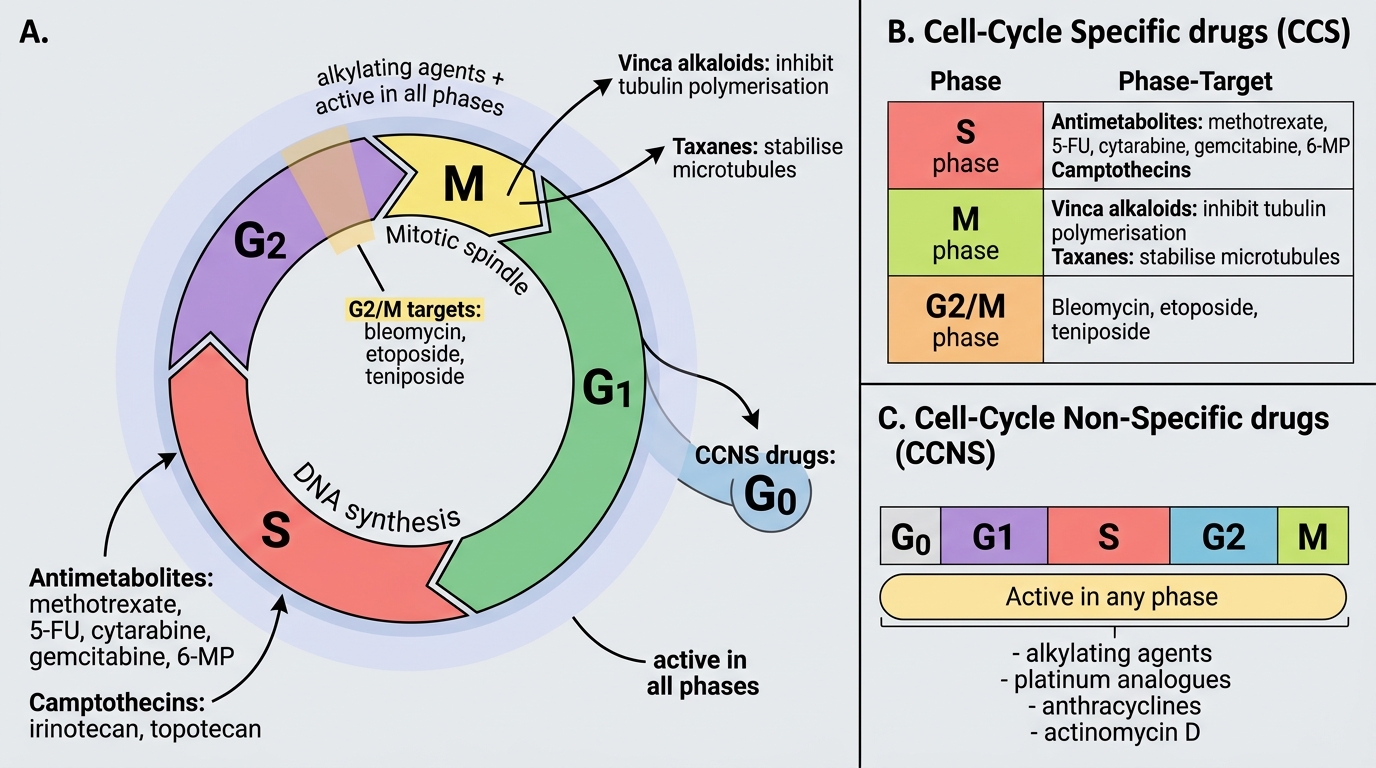
Cell-Cycle Targets of Anticancer Drugs
Classification: CCS vs CCNS, and Drug Class Taxonomy
Cell-cycle specificity is a key pharmacological property that determines when during cell division a drug is active:
Cell-Cycle Non-Specific (CCNS) — active in any phase including G0:
- Alkylating agents: Cyclophosphamide, ifosfamide, melphalan, chlorambucil, busulfan, carmustine (BCNU), dacarbazine; cisplatin, carboplatin, oxaliplatin (platinum analogues with alkylating-like mechanism)
- Anthracyclines: Doxorubicin, daunorubicin, epirubicin, idarubicin — primarily CCNS (though with G2/M preference)
- Actinomycin D (dactinomycin): Intercalates DNA
Cell-Cycle Specific (CCS) — active primarily in one cell-cycle phase:
- S-phase: Antimetabolites (methotrexate, 5-FU, cytarabine, gemcitabine, 6-MP, cladribine); camptothecins (irinotecan, topotecan — topoisomerase I)
- M-phase (mitotic spindle disruption): Vinca alkaloids (vincristine, vinblastine — INHIBIT tubulin polymerisation → no spindle forms); Taxanes (paclitaxel, docetaxel — STABILISE microtubules → spindle cannot depolymerise — OPPOSITE mechanism to vincas)
- G2/M-phase: Bleomycin (DNA strand scission via free radicals); Etoposide, teniposide (topoisomerase II inhibitors)
Drug class taxonomy:
1. Alkylating agents (DNA crosslinks → double-strand breaks → CCNS)
2. Antimetabolites (false nucleotide precursors → S-phase arrest)
3. Plant alkaloids: (a) Vinca alkaloids — antimitotic; (b) Taxanes — antimitotic opposite mechanism; (c) Topoisomerase inhibitors — etoposide (II), irinotecan/topotecan (I)
4. Antibiotics (antibiotic structure, anticancer use): Anthracyclines (doxorubicin); Bleomycin; Actinomycin D; Mitomycin C
5. Hormonal/endocrine agents: Tamoxifen (ER antagonist); LHRH analogues; Aromatase inhibitors; Steroids
6. Targeted/biological agents: Tyrosine kinase inhibitors (imatinib — BCR-ABL); Monoclonal antibodies (trastuzumab — HER2; rituximab — CD20; bevacizumab — VEGF)