Page 37 of 46
PH8.11 | PH8.11 | Anticancer Drugs and Toxicity Amelioration — SDL Guide — SDL Guide (Part 2)
Alkylating Agents and Antimetabolites: Mechanisms, Uses, and Toxicities
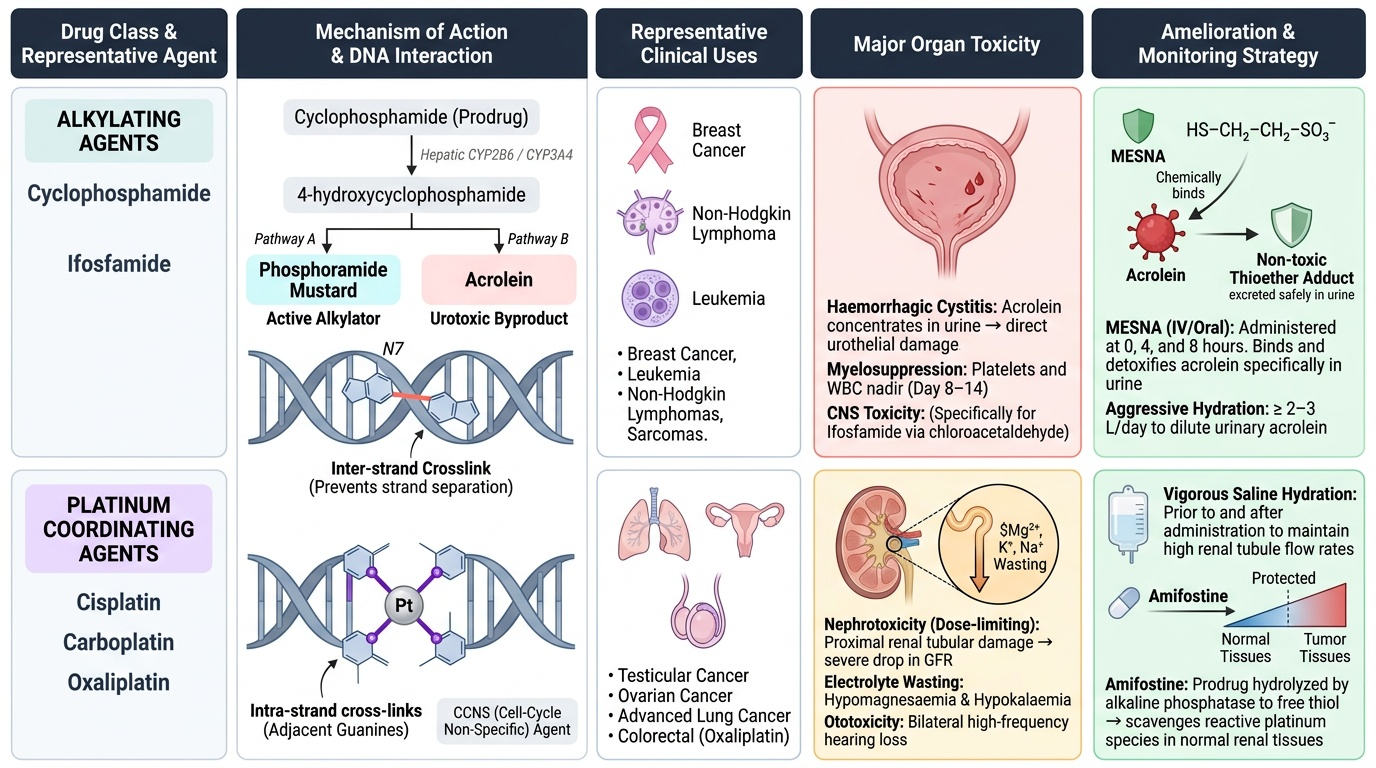
Alkylating Agents — DNA damage by covalent crosslinks:
Alkylating agents are electrophilic compounds that form reactive carbonium ions (or related species) → covalent bonds with nucleophilic sites on DNA, particularly N7 of guanine. These adducts cause: inter-strand crosslinks (prevent DNA strand separation → replication failure); intra-strand crosslinks; DNA-protein crosslinks; all leading to apoptosis. Because this damage is not cell-cycle-dependent (DNA exists and can be alkylated in all phases), these are CCNS agents — effective against quiescent tumour cells.
Cyclophosphamide: Prodrug — hepatic CYP2B6/CYP3A4 hydroxylation → 4-hydroxycyclophosphamide → phosphoramide mustard (active alkylator) + acrolein (urotoxic byproduct). Acrolein is concentrated in the urinary bladder → direct urothelial damage → haemorrhagic cystitis (dysuria, haematuria, potentially fatal bladder bleeding). Amelioration: MESNA (sodium 2-mercaptoethane sulfonate) — reacts with acrolein in the urine (not in blood/tumour) → forms a non-toxic thioether adduct → detoxifies acrolein specifically in the urinary tract. Adequate hydration (≥2–3L/day) dilutes acrolein. MESNA given IV at 0, 4, and 8 hours after each cyclophosphamide dose. Also: ifosfamide similarly requires MESNA (produces acrolein + chloroacetaldehyde — additional CNS toxicity).
Other systemic cyclophosphamide toxicities: severe myelosuppression (nadir day 8–14); nausea/vomiting; alopecia; SIADH (syndrome of inappropriate ADH — hyponatraemia); sterility.
Cisplatin (and carboplatin, oxaliplatin): Platinum compounds form platinum-DNA adducts — predominantly intra-strand cross-links at adjacent guanines — functionally similar to bifunctional alkylation. CCNS. Cisplatin toxicity profile:
- Nephrotoxicity (dose-limiting): proximal renal tubular damage → reduced GFR, electrolyte loss (Mg²⁺, K⁺, Na⁺ wasting — hypomagnesaemia can persist months after stopping cisplatin). Amelioration: aggressive IV hydration (1–3L normal saline before and after each cisplatin dose — the most important protection); amifostine (prodrug converted by alkaline phosphatase on kidney/normal tissue surfaces to free thiol → scavenges platinum adducts preferentially in normal tissues vs tumour tissue). Carboplatin is less nephrotoxic than cisplatin (different leaving group; slower platinum-DNA adduct formation).
- Ototoxicity (irreversible): cumulative cochlear hair cell damage → high-frequency hearing loss (tinnitus → permanent); audiometry monitoring is essential in paediatric patients. No effective ameliorant for established ototoxicity — prevention = avoid ototoxic combinations (aminoglycosides + cisplatin is especially dangerous).
- Severe CINV (chemotherapy-induced nausea/vomiting): cisplatin is the most emetogenic chemotherapy agent — requires aggressive anti-emetic prophylaxis: 5-HT₃ antagonist (ondansetron) + NK1 receptor antagonist (aprepitant/fosaprepitant) + dexamethasone triple combination.
- Peripheral neuropathy: sensory > motor; dose-cumulative; can be severe and persistent.
Methotrexate (MTX): Antimetabolite — competitive inhibitor of dihydrofolate reductase (DHFR) → blocks DHF → THF conversion → depletes thymidylate (via thymidylate synthase) and purines → DNA synthesis blocked → S-phase arrest. CCS. Used at low doses for rheumatoid arthritis, psoriasis (folate pathway inhibition is also its mechanism here, with DHFR the primary target). At high doses (HD-MTX) for osteosarcoma, CNS lymphoma, leukaemia. High-dose MTX toxicity (mucositis, nephrotoxicity, myelosuppression) is managed by Leucovorin (folinic acid, 5-formyl-THF) rescue: leucovorin is reduced folate that bypasses DHFR → rescues normal tissues from MTX effect (tumour cells are less dependent on leucovorin rescue due to different folate transporter kinetics and intracellular MTX polyglutamation). Leucovorin rescue is timed to start 24 hours after HD-MTX infusion ends. MTX levels are measured serially — leucovorin dose and duration adjusted until MTX clears.
5-Fluorouracil (5-FU): Fluorinated pyrimidine analogue — prodrug converted intracellularly to FdUMP (inhibits thymidylate synthase → blocks thymidylate → DNA synthesis); also incorporated into RNA (disrupts RNA processing). CCS. Leucovorin (folinic acid) modulates 5-FU by stabilising the ternary complex (FdUMP-TS-leucovorin) → prolongs TS inhibition → enhances 5-FU activity. Toxicities: mucositis (dose-limiting for IV), hand-foot syndrome (palmar-plantar erythrodysaesthesia — for capecitabine, the oral prodrug), myelosuppression, rare cardiotoxicity (coronary artery spasm).
6-Mercaptopurine (6-MP) and 6-Thioguanine: Purine analogues — activated by HPRT (hypoxanthine-guanine phosphoribosyltransferase) → thioguanine nucleotides → incorporated into DNA → S-phase arrest. Critical drug interaction: allopurinol (xanthine oxidase inhibitor, used for hyperuricaemia in tumour lysis) inhibits the catabolism of 6-MP by xanthine oxidase → 4–5× accumulation of 6-MP → severe myelosuppression. If allopurinol must be co-administered, reduce 6-MP to 25–33% of normal dose. TPMT (thiopurine methyltransferase) polymorphism: TPMT methylates 6-MP → inactive metabolite. TPMT-deficient patients (autosomal recessive; ~1:300 persons of European ancestry) cannot methylate 6-MP → accumulate toxic thioguanine nucleotides → profound myelosuppression at standard doses. Screening for TPMT activity or genotype before starting 6-MP/azathioprine is recommended.
Provided image
SELF-CHECK
A patient receiving cyclophosphamide for lymphoma develops frank haematuria on day 3 of treatment. MESNA was not administered. What is the mechanism of this adverse effect and what should have been done to prevent it?
A. Cyclophosphamide directly alkylates bladder urothelium → haemorrhagic cystitis; no prevention possible with standard dosing
B. Acrolein, a urotoxic metabolite of cyclophosphamide, damages bladder epithelium; MESNA (2-mercaptoethane sulfonate) reacts with acrolein in urine → detoxification; should have been given with each cyclophosphamide dose
C. Immune-mediated haemorrhagic cystitis from cyclophosphamide; prevented by corticosteroids
D. Nephrotoxic renal tubular damage from cyclophosphamide spilling into the bladder; prevented by aggressive IV hydration
Reveal Answer
Answer: B. Acrolein, a urotoxic metabolite of cyclophosphamide, damages bladder epithelium; MESNA (2-mercaptoethane sulfonate) reacts with acrolein in urine → detoxification; should have been given with each cyclophosphamide dose
Cyclophosphamide is hepatically activated to phosphoramide mustard (active alkylating agent) and acrolein (the nephrotoxic/urotoxic byproduct). Acrolein is concentrated in urine in the bladder → direct chemical urothelial damage → haemorrhagic cystitis (dysuria, haematuria, bladder wall fibrosis in severe cases). MESNA (sodium 2-mercaptoethane sulfonate) is specifically designed to react with acrolein in the urinary tract (urinary thiol scavenging) → forms non-toxic thioether adduct → completely prevents acrolein-mediated bladder damage. MESNA must be administered with each cyclophosphamide dose (typically IV at 0h, 4h, 8h post-cyclophosphamide). Adequate hydration (≥2–3L/day) is an essential additional protective measure. The bladder damage is not immune-mediated, and is not nephrotoxic in mechanism.
Plant Alkaloids, Antibiotics, and Targeted Agents
Vinca Alkaloids (vincristine, vinblastine, vinorelbine):
- Mechanism: bind β-tubulin → INHIBIT tubulin polymerisation → prevent formation of the mitotic spindle → metaphase arrest → CCS (M-phase). At low concentrations: induce microtubule depolymerisation; at higher concentrations: prevent new spindle microtubule polymerisation.
- Vincristine: used in leukaemias (ALL), lymphomas; NOT myelosuppressive (unique among cytotoxic drugs — this is why vincristine is combined with other myelosuppressive agents without bone marrow dose limitations). This is a key pharmacological fact for combination regimen design.
- Toxicities: Peripheral neuropathy (dose-limiting for vincristine — sensory > motor > autonomic; areflexia, paraesthesia, foot drop in severe cases; autonomic: constipation from reduced gut motility; paralytic ileus — prevent with laxatives/stool softeners from day 1 of vincristine). Alopecia. SIADH (rare). Inadvertent intrathecal injection of vincristine is invariably fatal — always labelled clearly and administered only IV.
- Vinblastine: more myelosuppressive than vincristine; used in Hodgkin lymphoma (ABVD regimen), testicular cancer. Less neurotoxic than vincristine.
Taxanes (paclitaxel, docetaxel):
- Mechanism: bind β-tubulin → STABILISE microtubules → prevent depolymerisation → spindle cannot disassemble → cells cannot complete mitosis or cytokinesis → CCS (M-phase). Opposite mechanism to vinca alkaloids — vincas prevent spindle FORMATION; taxanes prevent spindle DISASSEMBLY. Both result in mitotic arrest, but the molecular mechanism is entirely different.
- Paclitaxel: dissolved in Cremophor EL (polyoxyethylated castor oil) → severe hypersensitivity reactions (anaphylaxis risk); premedicate with dexamethasone + diphenhydramine + H2-blocker. Myelosuppression (neutropenia), peripheral neuropathy (sensory), alopecia, bradycardia (cardiac conduction delay).
- Docetaxel: dissolved in polysorbate 80 (less hypersensitivity than paclitaxel); fluid retention syndrome (pleural effusions, oedema — prevent with dexamethasone). Used in breast, lung, prostate cancers.
Bleomycin:
- Mechanism: bleomycin-Fe²⁺ complex binds DNA → iron is oxidised → electrons transferred to O₂ → reactive oxygen species (free radicals) → DNA strand scission (single and double strand breaks). G2/M-phase sensitive. No myelosuppression — unique (can be added to myelosuppressive regimens).
- Toxicities: Pulmonary toxicity (the primary dose-limiting toxicity) — bleomycin accumulates in the lungs (lack pulmonary bleomycin hydrolase, which inactivates bleomycin in other tissues). Progressive interstitial pneumonitis → fibrosis → irreversible if not caught early. Monitor: baseline and periodic pulmonary function tests (DLCO — diffusing capacity for CO — the most sensitive parameter; falls before clinical symptoms). Dose limit: avoid exceeding 400 units cumulative dose. Perioperative oxygen hazard: high FiO₂ in bleomycin-exposed patients can precipitate acute pulmonary damage (free-radical generation enhanced by hyperoxia). Anaesthesiologists must be informed of prior bleomycin treatment — use minimum effective FiO₂ (aim ≤35% where safe). Skin toxicity: hyperpigmentation, erythema, thickening (particularly of pressure areas). No alopecia (unlike most cytotoxics).
Doxorubicin (anthracycline):
- Mechanism: (1) DNA intercalation (inserts between base pairs → structural deformation → transcription/replication failure); (2) Topoisomerase II inhibition (stabilises topoisomerase II-DNA cleavage complex → permanent DNA strand breaks); (3) Free radical generation (quinone group of doxorubicin → semiquinone radical → O₂ → reactive oxygen species → cell membrane lipid peroxidation). The combination of these mechanisms accounts for its broad-spectrum anticancer activity.
- Cardiotoxicity (cumulative, dose-dependent): free radical generation in cardiomyocytes (which are relatively deficient in superoxide dismutase and catalase compared to dividing tumour cells) → progressive oxidative damage → dilated cardiomyopathy. Risk increases markedly above lifetime cumulative doses of 450–550 mg/m² for doxorubicin (lower thresholds for epirubicin ≥900 mg/m² is typically safe but higher than doxorubicin equivalent). Monitoring: echocardiography (left ventricular ejection fraction, LVEF) before treatment and after every 2–3 cycles at high cumulative doses. Dexrazoxane — an EDTA analogue that chelates iron → reduces intramyocardial free iron available for anthracycline-mediated free radical generation → cardioprotection. Used when cumulative doses approach the threshold or when there are existing cardiac risk factors.
- Other ADRs: severe myelosuppression (nadir day 10–14); mucositis; alopecia; red discolouration of urine (harmless — warn patient); vesicant (causes severe tissue necrosis if extravasated — must be given through a secure IV, not peripheral if possible).
Targeted agents (selected):
- Imatinib (Gleevec/Glivec) — selective BCR-ABL tyrosine kinase inhibitor (TKI); binds the ATP-binding site of BCR-ABL fusion kinase (constitutively active in CML — chronic myeloid leukaemia); also inhibits c-KIT (GIST) and PDGFR. Transformed CML from an incurable disease (median survival <5 years pre-imatinib) to a manageable chronic condition (10-year survival >80%). IRIS trial demonstrated superiority to interferon + cytarabine.
- Trastuzumab (Herceptin) — humanised monoclonal antibody against HER2 (ErbB2) receptor → inhibits HER2 signalling + ADCC; used in HER2-amplified breast and gastric cancers. Cardiotoxicity (reversible, unlike anthracyclines; manageable by withholding trastuzumab; synergistic cardiotoxicity with anthracyclines — avoid concurrent use when possible).
- Rituximab — chimeric anti-CD20 monoclonal antibody; depletes B-lymphocytes via ADCC + CDC + direct apoptosis; used in B-cell lymphomas (DLBCL), CLL, autoimmune diseases. Infusion reactions (premedicate); progressive multifocal leukoencephalopathy (PML — rare, due to JC virus reactivation in B-cell depleted patients).
SELF-CHECK
A patient with Hodgkin lymphoma receives ABVD chemotherapy (doxorubicin + bleomycin + vinblastine + dacarbazine). Three cycles later, he needs an elective inguinal hernia repair under general anaesthesia. What should the anaesthesiologist know about his chemotherapy regimen and what precaution is required?
A. Doxorubicin causes intraoperative cardiac arrhythmias — avoid volatile anaesthetic agents and use total IV anaesthesia
B. Bleomycin sensitises the lungs to oxygen-mediated injury — use the lowest effective FiO₂ (aim ≤35%), avoid prolonged high-concentration oxygen supplementation
C. Vinblastine causes severe myelosuppression — check CBC and postpone surgery if neutropenia is present
D. Dacarbazine is hepatically metabolised — avoid suxamethonium as both compete for hepatic CYP enzymes
Reveal Answer
Answer: B. Bleomycin sensitises the lungs to oxygen-mediated injury — use the lowest effective FiO₂ (aim ≤35%), avoid prolonged high-concentration oxygen supplementation
Bleomycin accumulates in the lungs (where it is poorly inactivated, unlike other tissues that have bleomycin hydrolase). It causes free-radical-mediated pulmonary endothelial and epithelial damage. High oxygen concentrations (high FiO₂) dramatically exacerbate bleomycin-induced pulmonary toxicity by enhancing free radical generation — even months to years after bleomycin treatment has ended. This is a well-documented anaesthetic risk: multiple reports of fatal post-operative pulmonary deterioration in bleomycin-exposed patients given high intraoperative oxygen. The anaesthesiologist must: (1) be informed of bleomycin exposure (past or present); (2) use the minimum effective FiO₂ (aim ≤30–35% if SpO₂ is maintained); (3) avoid PEEP settings that would require increased FiO₂; (4) consider CPAP/BiPAP rather than invasive ventilation where possible post-operatively. Vinblastine's myelosuppression is a valid concern but not the primary anaesthetic consideration. The others are incorrect.
Toxicity Amelioration: Organ-Specific Protection Strategies
PH8.11 explicitly requires a plan for amelioration of anticancer drug-induced toxicities. The following toxicity-amelioration framework covers the most common and clinically important organ toxicities:
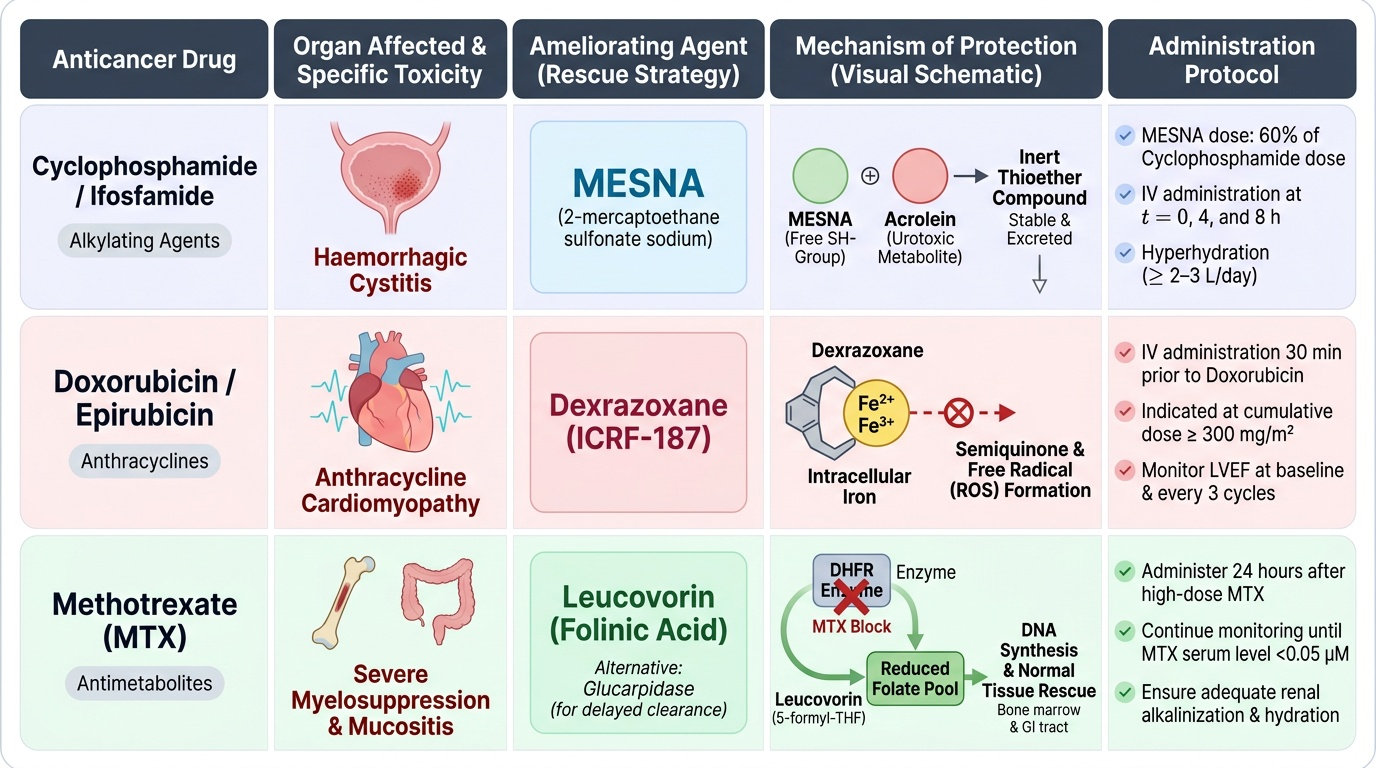
1. Haemorrhagic Cystitis — Cyclophosphamide/Ifosfamide:
- Agent: MESNA (2-mercaptoethane sulfonate sodium)
- Mechanism: Free SH group in MESNA reacts with acrolein (the urotoxic metabolite) in urine → inert thioether compound. MESNA is stable in blood but activated in urine — selective urothelial protection without systemic anti-tumour effect.
- Protocol: MESNA 60% of cyclophosphamide dose, given IV at 0, 4, and 8 hours (or continuous infusion for high-dose regimens). Also: adequate IV hydration (≥2–3L/day), bladder irrigation for high-dose regimens.
2. Anthracycline Cardiomyopathy — Doxorubicin/Epirubicin:
- Agent: Dexrazoxane (ICRF-187)
- Mechanism: Dexrazoxane is an EDTA analogue that chelates intracellular iron → reduces the availability of iron to participate in anthracycline-mediated semiquinone radical formation → reduced oxidative cardiomyocyte damage. Does NOT reduce anti-tumour efficacy of doxorubicin at standard clinical doses.
- Protocol: Administer IV 30 minutes before doxorubicin; used when cumulative doxorubicin dose approaches 300 mg/m² or when cardiac risk factors are present. Also: monitoring LVEF by echo before cycle 1 and every 3 cycles; cumulative dose limits.
3. High-Dose Methotrexate Toxicity — Leucovorin Rescue:
- Agent: Leucovorin (folinic acid, 5-formyl-THF) / alternatively Glucarpidase (enzyme that cleaves MTX — used for delayed MTX clearance)
- Mechanism: Leucovorin is a reduced folate that enters the folate cycle downstream of DHFR → bypasses MTX block → rescues normal tissues (bone marrow, GI mucosa) from MTX toxicity. Tumour cells preferentially retain MTX (due to polyglutamation) and are less rescued.
- Protocol: Start leucovorin 24–36 hours after MTX infusion ends; continue every 6 hours until serum MTX falls to <0.05–0.1 μM. Monitor serum MTX levels and creatinine — delayed MTX clearance (renal impairment) requires higher leucovorin doses and longer duration.
4. Cisplatin Nephrotoxicity:
- Strategies: (a) IV hydration (1–3L normal saline before + 1–2L after cisplatin infusion); (b) Amifostine (WR-2721) — aminothiol prodrug converted by alkaline phosphatase (higher activity in normal tissues than tumours) to active thiol → scavenges platinum adducts selectively in normal tissues; (c) Dose reduction/switch to carboplatin in patients with pre-existing renal impairment; (d) Avoid concurrent nephrotoxic drugs (aminoglycosides, NSAIDs); (e) Monitor creatinine and electrolytes (Mg²⁺ particularly); supplement Mg²⁺ (cisplatin-induced hypomagnesaemia is common and persistent).
5. Chemotherapy-Induced Nausea and Vomiting (CINV):
- High emetogenicity (cisplatin): triple antiemetic prophylaxis — ondansetron (5-HT₃ antagonist) + aprepitant/fosaprepitant (NK1 receptor antagonist) + dexamethasone. This three-drug combination reduces acute and delayed CINV by >80%.
- Moderate emetogenicity (doxorubicin, cyclophosphamide): ondansetron + dexamethasone ± aprepitant.
- Low emetogenicity (vinca alkaloids, taxanes): metoclopramide or ondansetron PRN.
- NK1 receptor antagonist mechanism: aprepitant blocks substance P (NK1 receptor in the brainstem vomiting centre) → reduces delayed CINV (day 2–5 after cisplatin, which is primarily mediated by substance P, not 5-HT₃).
6. Myelosuppression — Granulocyte Colony-Stimulating Factor (G-CSF):
- G-CSF (filgrastim, pegfilgrastim) stimulates granulopoiesis → reduces depth and duration of neutropenia → reduces risk of febrile neutropenia. Indications: patients with >20% risk of febrile neutropenia per chemotherapy cycle; secondary prophylaxis after prior febrile neutropenia episode.
- Erythropoiesis-stimulating agents (EPO, darbepoetin) for chemotherapy-related anaemia — reduce transfusion need but potential thromboembolism and tumour-stimulation risks limit use.
7. Tumour Lysis Syndrome (TLS):
- Risk: bulky haematological malignancies (Burkitt's lymphoma, ALL) → massive cell death → hyperuricaemia, hyperkalaemia, hyperphosphataemia, hypocalcaemia → cardiac arrhythmia and renal failure.
- Prophylaxis: allopurinol (XO inhibitor → reduces urate production — FIRST LINE in low-risk TLS; given 24–48 hours before chemotherapy); rasburicase (recombinant urate oxidase → converts urate to allantoin — more soluble; preferred for high-risk TLS: rapid urate lowering). Note: rasburicase is contraindicated in G6PD deficiency (generates H₂O₂ → oxidative haemolysis in G6PD-deficient RBCs). Aggressive IV hydration + urine alkalinisation + cardiac monitoring.
8. Vinca alkaloid-induced constipation: Prophylactic laxatives/stool softeners from day 1 of vincristine; paralytic ileus risk in elderly or high-dose patients — early dietary and pharmacological bowel management.
Provided image
SELF-CHECK
A patient with Burkitt's lymphoma (massive retroperitoneal and mesenteric nodal disease) is started on chemotherapy. Within 12 hours, serum uric acid rises to 14 mg/dL, potassium 6.8 mEq/L, phosphate 7.0 mg/dL, creatinine 2.5 mg/dL. What drug is the most appropriate treatment for the hyperuricaemia, and what contraindication must be checked first?
A. Allopurinol — first-line for all cases of tumour lysis syndrome; no important contraindications
B. Rasburicase — converts uric acid to allantoin (rapidly reduces uric acid); check G6PD status first (contraindicated in G6PD deficiency)
C. Febuxostat — superior to allopurinol for TLS and has no relevant contraindications
D. Sodium bicarbonate IV — alkalinises urine to dissolve urate crystals; avoids the need for specific uricolytic drugs
Reveal Answer
Answer: B. Rasburicase — converts uric acid to allantoin (rapidly reduces uric acid); check G6PD status first (contraindicated in G6PD deficiency)
This patient has established tumour lysis syndrome (TLS) with high serum uric acid (14 mg/dL), hyperkalaemia, hyperphosphataemia, and acute kidney injury. Rasburicase (recombinant urate oxidase) is indicated for established/high-risk TLS — it directly converts uric acid to allantoin (more soluble; renal clearance enhanced) and reduces serum uric acid within hours, far faster than allopurinol (which only prevents new urate formation but does not reduce existing uric acid). Critical contraindication: rasburicase generates hydrogen peroxide (H₂O₂) as a byproduct. In G6PD-deficient patients, H₂O₂ cannot be neutralised → severe haemolytic anaemia. G6PD status must be checked (or G6PD-deficient patients presumed and rasburicase avoided) before administering rasburicase. In G6PD deficiency, use allopurinol with aggressive hydration instead. Febuxostat is another XO inhibitor but has less evidence and is not preferred for acute TLS. Sodium bicarbonate alkalinisation helps prevent urate crystallisation in the tubules but does not reduce uric acid levels.