Page 5 of 46
PH8.3 | PH8.3 | Antibacterial Drug Classes — SDL Guide — SDL Guide
Learning Objectives
- Explain the kinetics, dynamics, adverse effects, and indications of sulfonamides, quinolones, beta-lactams, macrolides, tetracyclines, and aminoglycosides
- Classify antibacterial drugs by their mechanism of action and target site
- Identify key adverse effects and contraindications for each major class
- Apply drug class knowledge to clinical prescribing scenarios
INSTRUCTIONS
Antibacterial pharmacology is the largest and most clinically essential chapter in your pharmacology curriculum. You will encounter bacterial infections in every specialty of medicine — and the choice of antibiotic determines patient outcomes, resistance patterns, and institutional antimicrobial ecology. This module covers the six major classes mandated by NMC, plus key newer agents, building on the resistance and stewardship principles from the previous SDL.
References
- Tripathi KD. Essentials of Medical Pharmacology, 8th ed., Ch 56–62 (textbook)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, 13th ed., Ch 56–61 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 72-year-old man with diabetes and chronic kidney disease is admitted with urinary sepsis. His serum creatinine is 3.5 mg/dL (eGFR ~18 mL/min). Blood cultures grow Klebsiella pneumoniae — ESBL-positive, sensitive only to meropenem and amikacin. The attending wants to add amikacin. The pharmacist flags: 'Sir, at this eGFR, amikacin without monitoring will destroy his remaining kidney function.' The attending disagrees: 'He is septic, I need bactericidal coverage.' Who is right? How do you dose amikacin in severe renal impairment while still achieving therapeutic efficacy? And why is the first choice of antibacterial for ESBL sepsis not ceftriaxone, even though ESBL Klebsiella was once ceftriaxone-sensitive? The answers emerge from a thorough understanding of antibacterial pharmacokinetics, mechanisms, and the logic of resistance.
WHY THIS MATTERS
The six drug classes covered in this SDL — sulfonamides, quinolones, beta-lactams, macrolides, tetracyclines, and aminoglycosides — form the backbone of antimicrobial prescribing worldwide. In India, these drugs account for over 80% of antibiotic prescriptions. Each class has a unique target, spectrum, and toxicity profile. Understanding these at the mechanistic level — not as a memorised list but as a logical system — allows you to predict drug behaviour, recognise toxicities before they become severe, and choose appropriately across thousands of clinical situations you will face in your career.
RECALL
From SDL 1 of this cluster, recall: (1) selective toxicity — exploiting bacterial-specific structures; (2) bacteriostatic vs bactericidal — tetracyclines and macrolides are static; aminoglycosides are bactericidal despite being 30S inhibitors; (3) PK/PD indices — T>MIC governs β-lactam dosing; AUC/MIC and Cmax/MIC govern aminoglycosides and fluoroquinolones; (4) resistance mechanisms — β-lactamases, efflux pumps, PBP mutations, target methylation, porin loss.
From Year-1 Microbiology: gram-positive organisms lack an outer membrane but have thick peptidoglycan; gram-negative organisms have a thin peptidoglycan surrounded by an outer membrane (lipopolysaccharide layer) — this outer membrane limits drug entry and is why most gram-positives are naturally sensitive to penicillin while gram-negatives often are not.
The Bacterial Battleground: Pathophysiology of Bacterial Infection
Successful antibacterial therapy requires understanding the interaction between pathogen, drug, and host at three levels: the target-site concentration of the drug, the susceptibility of the organism, and the host's ability to eliminate bacteria once the drug has inhibited their growth or multiplication.
Bacteria cause disease through several mechanisms: direct invasion and tissue destruction (e.g. S. pyogenes producing streptokinase, hyaluronidase, and toxins); toxin production (exotoxins: S. aureus TSST-1, C. difficile TcdA/TcdB; endotoxin from gram-negative LPS causing septic shock); and competition for host resources with displacement of normal flora (C. difficile pseudomembranous colitis after antibiotic disruption of microbiome).
The site of infection matters because drugs must reach the focus in adequate concentration: meningitis requires a drug that crosses the blood-brain barrier (ceftriaxone, chloramphenicol, penicillin at high dose — but NOT aminoglycosides, which penetrate CSF poorly); intracellular infections (Mycobacteria, Chlamydia, Brucella, Legionella) require drugs that accumulate inside cells (azithromycin, doxycycline, fluoroquinolones — not β-lactams, which are extracellular agents); biofilms (Pseudomonas in cystic fibrosis lung, device-associated infections) dramatically reduce antibiotic efficacy and require combination regimens.
The pathogen identity drives class selection: gram-positive cocci (Staphylococcus, Streptococcus) remain sensitive to penicillinase-resistant penicillins (cloxacillin) unless MRSA; gram-negative enteric organisms require extended-spectrum agents; anaerobes (Bacteroides, Clostridium) are targeted by metronidazole, clindamycin, or broad-spectrum β-lactam/inhibitor combinations.
Therapeutic Goal: Targeting Bacterial Machinery Without Harming the Host
The therapeutic goal of antibacterial therapy can be stated precisely: achieve a drug concentration at the site of infection that exceeds the MIC for the pathogen, for long enough (time-dependent drugs) or at high enough a peak (concentration-dependent drugs) to achieve a clinical and microbiological cure — while keeping the concentration below the threshold for host toxicity. This balance defines the therapeutic window.
Each antibacterial class exploits a target site with some degree of bacterial specificity. The map of targets is also the map of classes: cell-wall synthesis (β-lactams, glycopeptides); 30S ribosomal subunit (aminoglycosides, tetracyclines); 50S ribosomal subunit (macrolides, chloramphenicol, clindamycin, linezolid); DNA replication machinery — specifically DNA gyrase (subunits GyrA/GyrB) and topoisomerase IV (ParC/ParE) — targeted by fluoroquinolones; folate biosynthesis pathway (sulfonamides inhibit dihydropteroate synthase; trimethoprim inhibits dihydrofolate reductase — sequential block produces synergistic bactericidal activity in the combination).
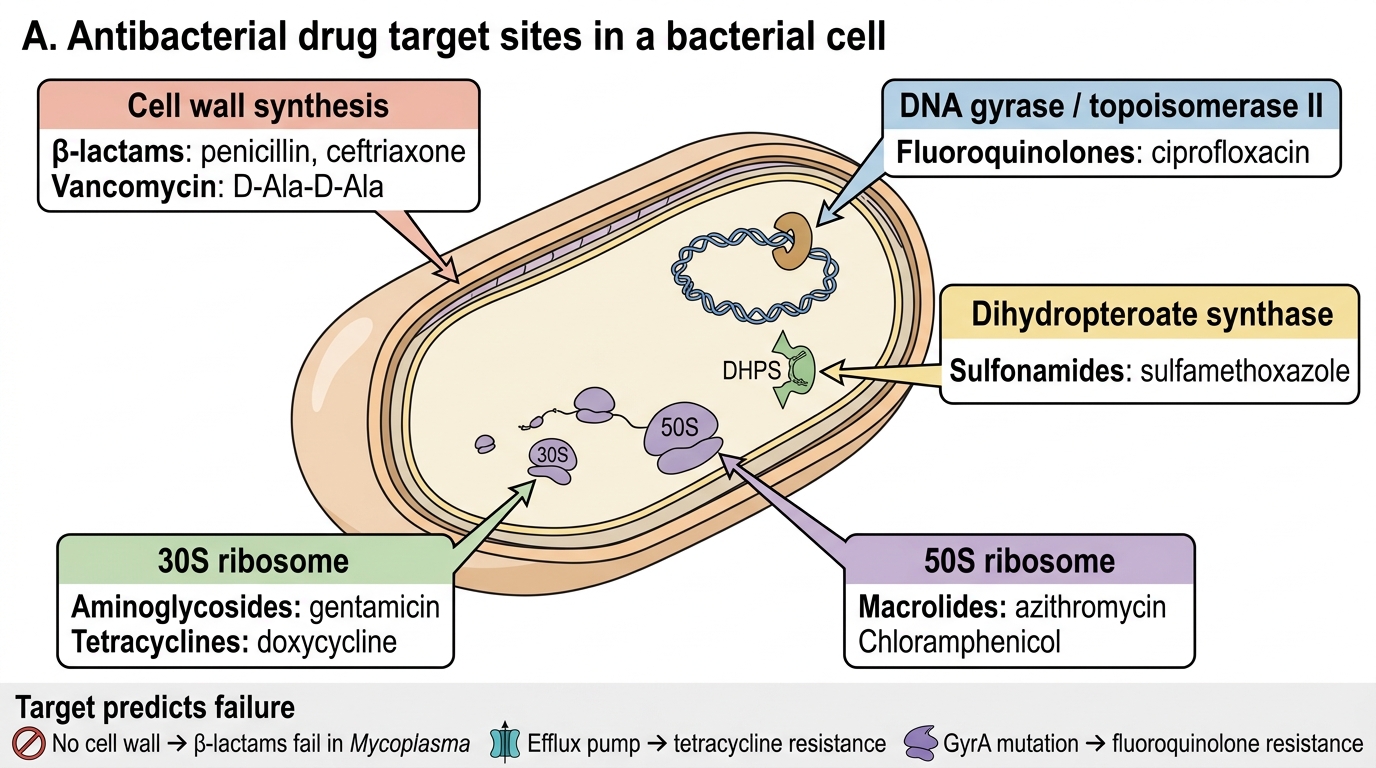
Antibacterial Drug Target Sites
Understanding the target also predicts the pattern of failure: a drug that works on cell-wall synthesis is ineffective against organisms without a cell wall (Mycoplasma — no peptidoglycan layer); tetracyclines fail against organisms with efflux pumps; fluoroquinolones fail when GyrA is mutated. These are not arbitrary resistance events — they are logical consequences of target biology.
Classification of Antibacterial Drugs by Target
A mechanistic classification is more useful than an alphabetical drug list because it allows prediction of cross-resistance, toxicity patterns, and spectrum from first principles. The five major target sites define the primary classes:
1. Cell-wall synthesis inhibitors:
- β-Lactams (penicillins, cephalosporins, carbapenems, monobactams) — inhibit PBPs (transpeptidases), preventing peptidoglycan cross-linking; bactericidal
- Glycopeptides (vancomycin, teicoplanin) — bind D-Ala-D-Ala termini of peptidoglycan precursors, blocking transglycosylation; bactericidal for gram-positives only (too large to cross gram-negative outer membrane)
- Fosfomycin — inhibits MurA (first step of peptidoglycan synthesis); bactericidal
2. 30S ribosomal inhibitors:
- Aminoglycosides (gentamicin, tobramycin, amikacin, streptomycin) — irreversible binding → misreading → membrane disruption → bactericidal
- Tetracyclines (tetracycline, doxycycline, minocycline) + tigecycline (glycylcycline) — reversible block of aminoacyl-tRNA binding → bacteriostatic
3. 50S ribosomal inhibitors:
- Macrolides (erythromycin, azithromycin, clarithromycin) — block peptide translocation → bacteriostatic
- Chloramphenicol — inhibits peptidyltransferase → bacteriostatic
- Clindamycin — inhibits peptide bond formation (similar binding site to macrolides, explaining cross-resistance — MLSB phenotype) → bacteriostatic
- Linezolid (oxazolidinone) — binds 23S rRNA of 50S subunit, blocks initiation complex formation → bacteriostatic against Staphylococcus/Enterococcus; may be bactericidal against Streptococcus
4. DNA replication/transcription inhibitors:
- Fluoroquinolones (ciprofloxacin, levofloxacin, moxifloxacin, norfloxacin) — inhibit DNA gyrase (gram-negatives: GyrA primary target) and topoisomerase IV (gram-positives: ParC primary target) → bactericidal, concentration-dependent
- Rifampicin — inhibits bacterial RNA polymerase (binds β-subunit) → bactericidal; NOT used alone (rapid single-step resistance)
- Metronidazole — prodrug activated by bacterial nitroreductases → reactive free radical damages DNA → bactericidal against strict anaerobes and microaerophiles
5. Folate pathway inhibitors:
- Sulfonamides — inhibit dihydropteroate synthase (competitive inhibitor of PABA) → bacteriostatic
- Trimethoprim — inhibits dihydrofolate reductase → bacteriostatic alone; bactericidal in TMP-SMX combination (sequential block, cotrimoxazole)