Page 1 of 36
PH7.1 | PH7.1 | Diabetes Mellitus Pharmacotherapy — SDL Guide — SDL Guide
Learning Objectives
- Classify antidiabetic drugs into insulin and non-insulin categories with subclass detail
- Describe the pharmacokinetics, pharmacodynamics, and adverse drug reactions of each drug class
- Apply clinical decision-making to devise a management plan for obese and non-obese diabetic patients
- Identify SGLT2 inhibitor eGFR thresholds for glycaemic versus cardio-renal indications
- Discuss strategies for prevention of diabetic complications including CV and renal endpoints
INSTRUCTIONS
Diabetes mellitus is among the most prevalent chronic diseases in India, affecting over 77 million adults. The pharmacological landscape has transformed dramatically with newer agent classes — GLP-1 receptor agonists, dual GIP/GLP-1 agonists, and SGLT2 inhibitors — that reduce not only blood glucose but also cardiovascular and renal events. This guide equips you to match the right drug to the right patient, moving beyond glucose-centricity to outcome-based prescribing.
References
- Tripathi KD. Essentials of Medical Pharmacology, 8th ed., Ch 19 (textbook)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, 13th ed., Ch 45 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 52-year-old obese software engineer (BMI 33, waist 102 cm) is diagnosed with Type 2 diabetes mellitus — HbA1c 8.9%, eGFR 61, with microalbuminuria, and a recent ECHO showing LVEF 42% (HFrEF). He asks: 'Doctor, will I need insulin? What are the safest tablets for my kidneys and heart?' You have a formulary with metformin, glibenclamide, sitagliptin, empagliflozin, and liraglutide. Which do you choose first, which do you add, and what thresholds matter? This guide builds the pharmacological logic to answer that question confidently.
WHY THIS MATTERS
India is the diabetes capital of the world. The disease shortens life by up to a decade through cardiovascular disease, diabetic nephropathy, retinopathy, and neuropathy. The choice of antidiabetic drug is no longer just about glucose control: SGLT2 inhibitors and GLP-1 RAs have demonstrated mortality reduction in patients with heart failure and CKD in landmark randomised trials. Understanding which drug to add — and knowing renal thresholds and contraindications — is a core clinical pharmacology competency (NMC PH7.1) that you will apply in every clinical posting.
RECALL
Before we examine drugs, anchor the physiology. In Type 2 diabetes mellitus (T2DM), two core defects co-exist: peripheral insulin resistance (principally in skeletal muscle and adipose tissue, mediated by impaired GLUT-4 signalling) and progressive β-cell dysfunction (declining insulin secretory capacity over years). The liver contributes via excess hepatic glucose output — driven by glucagon dominance and reduced insulin signalling. The result is both fasting hyperglycaemia (hepatic) and postprandial spikes (peripheral + β-cell failure). In Type 1 diabetes mellitus (T1DM), autoimmune destruction of β-cells produces absolute insulin deficiency from the outset — these patients require exogenous insulin for survival. Recall also the incretin system: gut L-cells release GLP-1 and K-cells release GIP in response to oral glucose, potentiating insulin release and suppressing glucagon — incretin effect is 50–70% of postprandial insulin secretion in normal physiology but blunted in T2DM.
Pathophysiology of Diabetes: Defect Drives Treatment
The pathophysiological heterogeneity of diabetes is the rationale for its pharmacological diversity. In T2DM, the ominous octet described by DeFronzo encompasses eight organs contributing to hyperglycaemia: β-cell failure, insulin resistance in muscle and liver, incretin deficiency, α-cell glucagon hypersecretion, increased renal glucose reabsorption (elevated SGLT2 expression), altered adipocyte lipolysis, hypothalamic dysregulation, and gut microbiome shifts. Each of these nodes corresponds to one or more drug targets — and explains why combination therapy is the rule rather than the exception for sustained glycaemic control. The progressive nature of β-cell decline means that a drug adequate at diagnosis may require augmentation within 3–5 years as endogenous insulin secretion wanes.
In T1DM, the defect is absolute and singular: no insulin production. Accordingly, exogenous insulin in a physiological pattern (basal-bolus) is the only strategy. Oral agents that depend on residual β-cell function (sulfonylureas, DPP-4i, GLP-1 RAs) are ineffective in T1DM. Conversely, SGLT2 inhibitors have emerging roles in T1DM (under specialist supervision) though the risk of euglycaemic diabetic ketoacidosis is a critical hazard.
From a pharmacological perspective, knowing the defect-to-target map lets you predict which drug is rationally suited to each patient: weight gain and insulin excess point toward insulin-sensitisers; β-cell burnout points toward insulin replacement; CV/renal comorbidity points toward SGLT2i/GLP-1 RA regardless of HbA1c baseline.
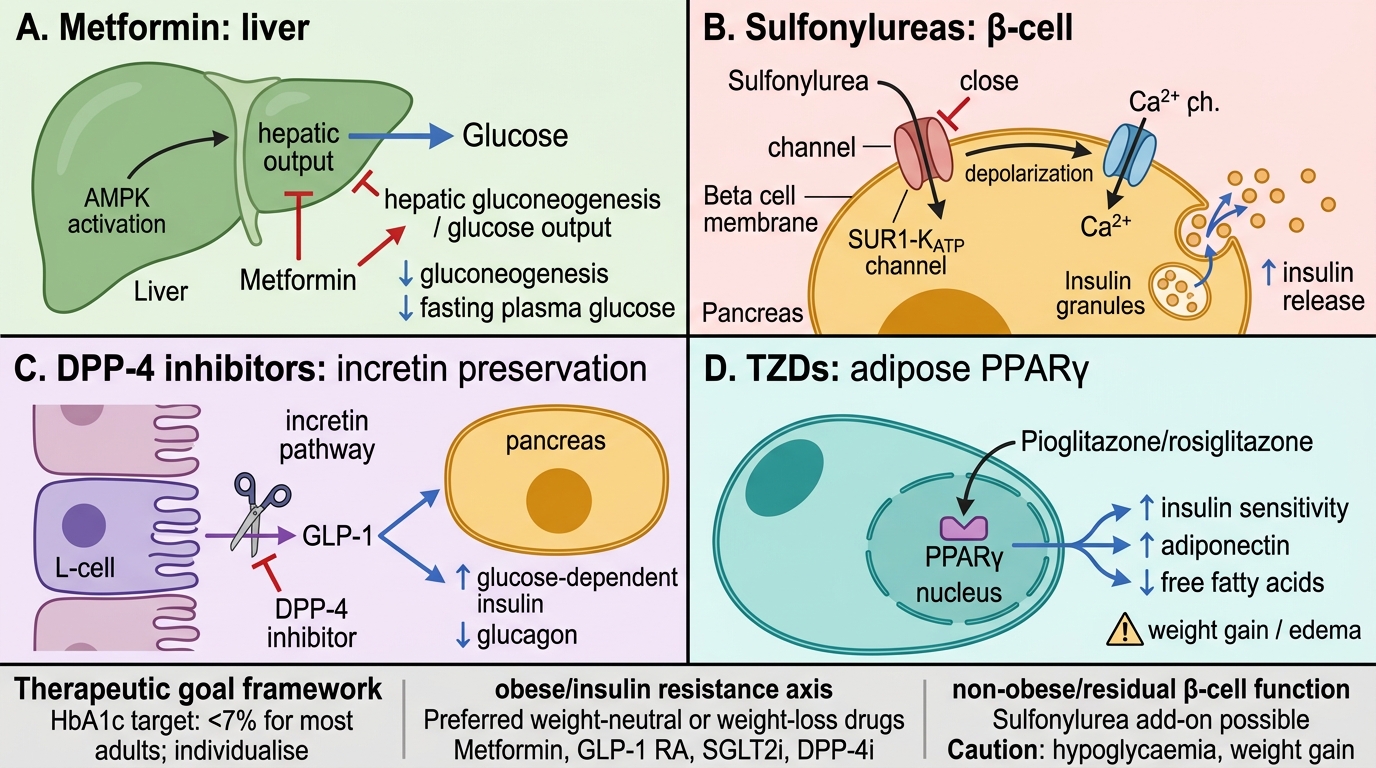
Mechanisms of Major Oral Antidiabetic Drugs
Therapeutic Goals and the Obese vs Non-Obese Framework
The central therapeutic goal in diabetes is preventing the microvascular and macrovascular complications that drive morbidity and mortality. The NMC and ADA define glycaemic targets as an HbA1c <7% for most adults with T2DM, with individualisation: stricter targets (<6.5%) for newly diagnosed, long-life-expectancy patients without hypoglycaemia risk; relaxed targets (<8%) for frail elderly, those with multiple comorbidities, or history of severe hypoglycaemia. Beyond glucose control, blood pressure <130/80 mmHg and LDL <70 mg/dL for high-CV-risk patients are co-equal goals.
The patient's body habitus is a primary decision axis. Obese patients (BMI ≥25 in Indians; ≥30 globally) have predominant insulin resistance — drugs that reduce weight or are weight-neutral are preferred: metformin (weight-neutral/slight reduction), GLP-1 RAs (3–5 kg loss), SGLT2 inhibitors (2–3 kg loss), DPP-4 inhibitors (weight-neutral). Insulin secretagogues (sulfonylureas, meglitinides) and insulin itself cause weight gain and are avoided unless necessary. Pioglitazone (TZD) also causes weight gain and fluid retention — low preference in obese patients. Non-obese patients with residual β-cell function can use sulfonylureas safely as add-on, since the weight-gain concern is less critical.
For patients with established cardiovascular disease, heart failure, or CKD, drug choice is now evidence-driven beyond glucose control: SGLT2 inhibitors and GLP-1 RAs have mortality/morbidity benefits proven in large RCTs and are recommended preferentially in guidelines regardless of HbA1c level.
| Patient Feature | Preferred Add-on (beyond metformin) | Avoid |
|---|---|---|
| Obesity | GLP-1 RA, SGLT2i, DPP-4i | SU, insulin, TZD |
| CVD/HF | SGLT2i, GLP-1 RA | TZD (HF worsening) |
| CKD (eGFR 20–45) | SGLT2i (cardio-renal dose), DPP-4i | Metformin (<30), SU |
| Hypoglycaemia risk | DPP-4i, GLP-1 RA, SGLT2i | SU, insulin (if avoidable) |
| Cost-constrained | SU, metformin | Newer agents |
SELF-CHECK
A 58-year-old T2DM patient with HbA1c 8.2%, eGFR 38, and recent LVEF 35% (HFrEF) is on metformin 500 mg BD. Which add-on is MOST appropriate?
A. A. Glibenclamide 5 mg
B. B. Pioglitazone 30 mg
C. C. Empagliflozin 10 mg
D. D. Sitagliptin 100 mg
Reveal Answer
Answer: C. C. Empagliflozin 10 mg
Empagliflozin (SGLT2i) is preferred in HFrEF and CKD. Its cardio-renal indication continues down to eGFR ~20 (EMPEROR-Reduced trial). Glibenclamide risks hypoglycaemia and is hepatically metabolised but accumulates in CKD. Pioglitazone is CONTRAINDICATED in HF (fluid retention worsens HFrEF). Sitagliptin is renal-safe (dose-reduced to 25–50 mg in CKD) and does not worsen HF, but lacks the mortality benefit empagliflozin has demonstrated.
Insulin: Types, Pharmacokinetics, and Clinical Use
Insulin remains the most potent glucose-lowering agent available and is essential in T1DM, during pregnancy, in acute metabolic decompensation, and as escalation therapy in T2DM with β-cell failure. Understanding the pharmacokinetic profile of each insulin formulation is critical to designing physiological replacement regimens.
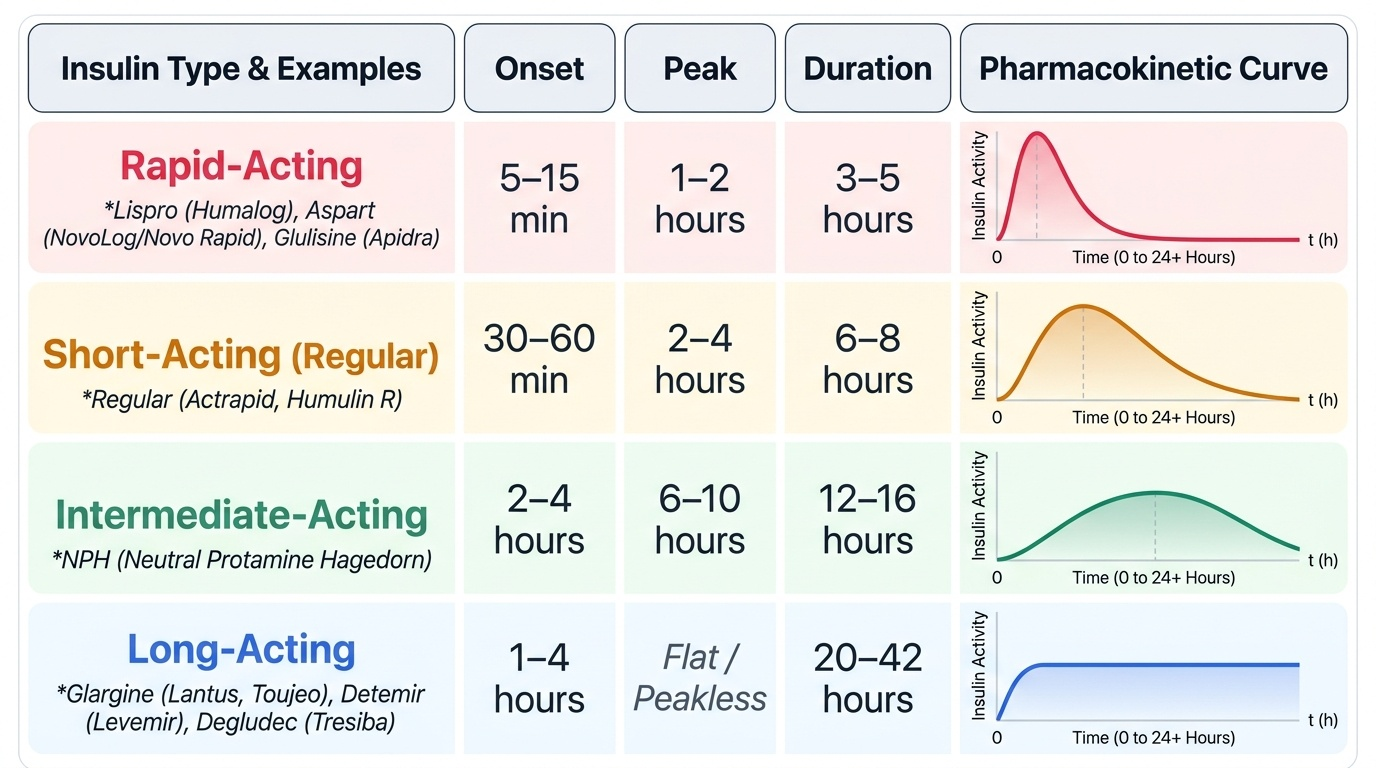
Rapid-acting analogues — insulin lispro (Humalog), insulin aspart (NovoLog/Novo Rapid), insulin glulisine (Apidra) — are produced by amino acid substitutions that reduce self-association, yielding onset within 5–15 minutes, peak at 1–2 hours, and duration of 3–5 hours. They are given immediately before or after a meal, closely mimicking the first-phase insulin response. Short-acting regular insulin (Actrapid, Humulin R) has slower subcutaneous absorption: onset 30–60 min, peak 2–4 hours, duration 6–8 hours — must be given 30 minutes before the meal.
Intermediate-acting NPH insulin (Neutral Protamine Hagedorn) has onset 2–4 hours, peak 6–10 hours, duration 12–16 hours; it requires twice-daily dosing and has an unpredictable peak that risks nocturnal hypoglycaemia. Long-acting analogues — insulin glargine (Lantus, Toujeo), insulin detemir (Levemir), insulin degludec (Tresiba) — provide near-peakless, steady basal coverage for 20–42 hours. Glargine is precipitated at the subcutaneous injection site (pH 4 solution becomes neutral in tissue), slowing absorption. Detemir binds albumin. Degludec forms multi-hexamer chains with ultra-long action (~42 hr) and the lowest pharmacodynamic variability — preferred in hypoglycaemia-prone patients.
The basal-bolus regimen — one long-acting insulin at night + rapid-acting analogues at each meal — most closely reproduces physiological insulin secretion and achieves the best HbA1c reduction (1.5–2.5%) with greatest flexibility. Premixed insulins (e.g. 30/70: 30% regular + 70% NPH) simplify administration but sacrifice individual titration.
Key ADRs: hypoglycaemia (most common), weight gain (0.5–1 kg per 1% HbA1c reduction), lipodystrophy at injection sites, local allergy (rare with modern human insulins).
Provided image