Page 18 of 36
PH7.6 | PH7.6 | Androgens and Erectile Dysfunction Drugs — SDL Guide — SDL Guide
Learning Objectives
- Classify androgen drugs by route of administration and describe the pharmacokinetics of testosterone preparations
- Explain the mechanism and clinical uses of 5α-reductase inhibitors and anti-androgens
- Describe the mechanism of action of PDE5 inhibitors and their critical contraindication with nitrates
- Devise a rational management approach for male hypogonadism, BPH, and erectile dysfunction
- Identify the adverse effects of anabolic steroid misuse
INSTRUCTIONS
Androgen pharmacology encompasses drugs at opposite ends of the therapeutic spectrum — from testosterone replacement for hypogonadism to anti-androgens for prostate cancer. The discovery of PDE5 inhibitors (sildenafil) revolutionised the treatment of erectile dysfunction and exemplifies how serendipity in pharmacology leads to major therapeutic advances. This guide covers both androgen pharmacology and the NO/cGMP-based mechanism of ED drugs, with a focus on the life-threatening nitrate-PDE5i contraindication.
References
- Tripathi KD. Essentials of Medical Pharmacology, 8th ed., Ch 21 (textbook)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, 13th ed., Ch 44 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 60-year-old man with known ischaemic heart disease on sublingual glyceryl trinitrate (GTN) as needed asks his general practitioner for sildenafil for erectile dysfunction. He is embarrassed about the ED but insists the medication is 'just a tablet.' His GP must explain why this combination is dangerous — not as a refusal, but as a safety-critical pharmacological interaction. Understanding the NO/cGMP mechanism of both drugs reveals exactly why their combination causes severe hypotension, and why the contraindication is absolute.
WHY THIS MATTERS
Erectile dysfunction affects approximately 150 million men worldwide and is increasingly recognised as a marker of cardiovascular disease. The pharmacology of androgens spans critical applications from childhood (delayed puberty, constitutional growth delay) through adulthood (male hypogonadism) to geriatric urology (BPH, prostate cancer). The PDE5 inhibitor-nitrate interaction is one of the most dangerous drug interactions in primary care — and one of the most frequently tested in clinical pharmacology examinations. Anti-androgens for prostate cancer represent a major pharmacological growth area in oncology.
RECALL
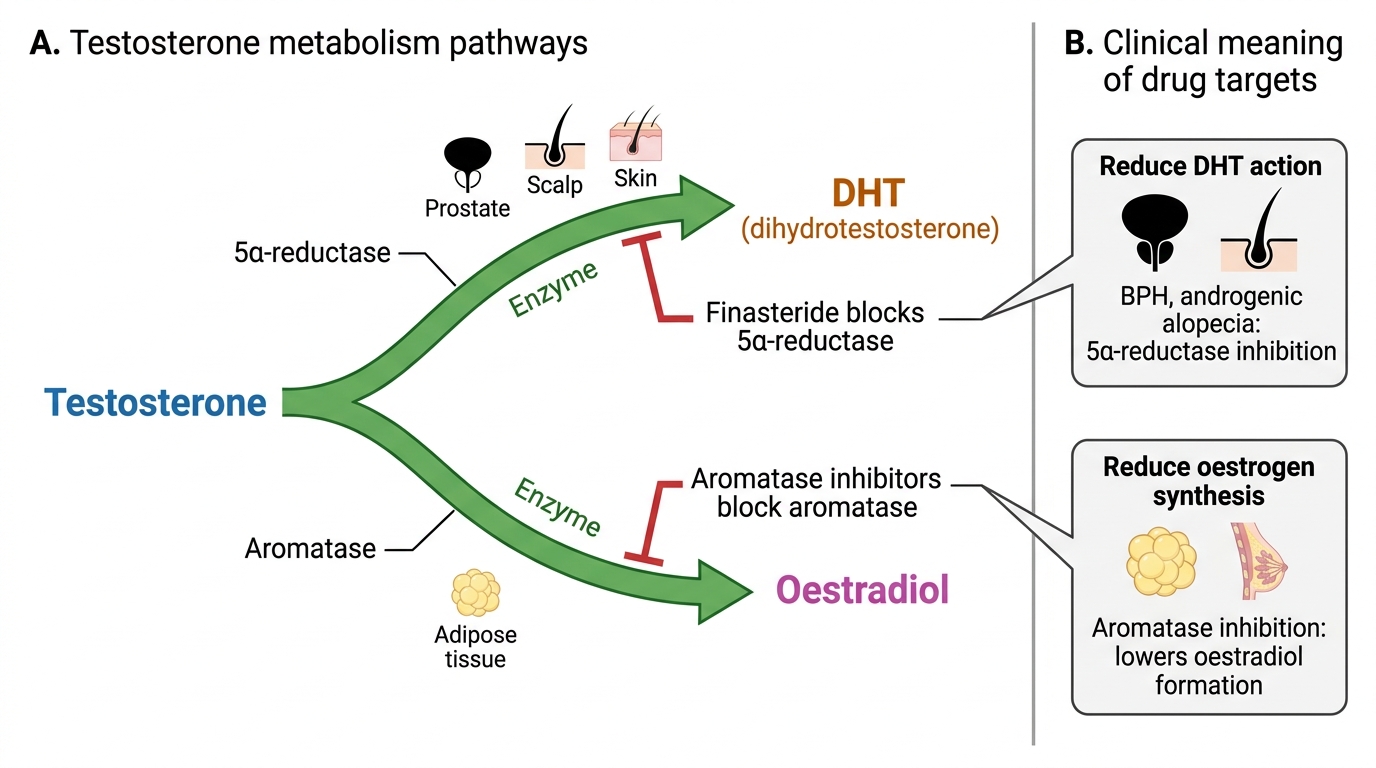
Androgen physiology: The HPG axis regulates testosterone production. LH from the anterior pituitary stimulates Leydig cells in the testicular interstitium to synthesise testosterone from cholesterol. FSH stimulates Sertoli cells to support spermatogenesis. Testosterone provides negative feedback on both the hypothalamus (reducing GnRH) and pituitary (reducing LH/FSH). Testosterone undergoes two critical peripheral metabolic conversions: (1) 5α-reductase in prostate, scalp, and skin converts testosterone to dihydrotestosterone (DHT) — the more potent androgen responsible for prostate growth (BPH, prostate cancer) and androgenetic alopecia; (2) aromatase in adipose tissue converts testosterone to oestradiol — explains gynaecomastia in conditions of androgen excess (adiposity, testicular tumours) and as an ADR of some androgens. Erectile dysfunction physiology: Sexual stimulation activates nonadrenergic noncholinergic (NANC) neurons in penile tissue → release of nitric oxide (NO) from penile endothelium → activates soluble guanylyl cyclase → increases cGMP in cavernosal smooth muscle → protein kinase G activation → Ca²⁺ channel inhibition → smooth muscle relaxation → lacunar spaces fill with blood → erection. PDE5 (phosphodiesterase type 5) hydrolyses cGMP, terminating the erection.
Androgen Physiology and Testosterone Metabolism
The physiological effects of androgens are mediated primarily by testosterone and its active metabolite DHT, both acting via the intracellular androgen receptor (AR) — a nuclear transcription factor. AR belongs to the steroid hormone receptor superfamily: testosterone/DHT enters the cell, binds cytoplasmic AR, the AR-androgen complex translocates to the nucleus and binds androgen response elements (AREs) in DNA, upregulating target gene transcription.
DHT versus testosterone: DHT binds the AR with 5× greater affinity than testosterone and has slower dissociation — making it the dominant intranuclear androgen in androgen-sensitive tissues (prostate, scalp, skin). In the hypothalamus and muscle, testosterone itself mediates effects. This tissue-specific metabolic conversion creates pharmacological opportunities: 5α-reductase inhibitors (finasteride, dutasteride) can selectively reduce DHT in the prostate/scalp without reducing testosterone in muscle and hypothalamus.
Physiological effects of testosterone:
- Male sexual differentiation (prenatal, external genitalia)
- Pubertal development (spermatogenesis, voice deepening, facial hair, penile/testicular growth, libido)
- Anabolic effects (muscle mass, bone density, nitrogen retention)
- Haematopoietic effects (stimulates erythropoietin → increased RBC production)
- Metabolic effects (reduces fat mass, increases muscle; complex cardiovascular effects)
The pharmacological corollaries are direct: testosterone replacement therapy in hypogonadism restores all these effects; anti-androgens in prostate cancer deprive cancer cells of the androgen signal they are addicted to.
Testosterone Metabolism and Drug Targets
Therapeutic Goals: Androgen Deficiency vs Androgen Excess
The therapeutic goals of androgen pharmacology are bifurcated: replace the deficient hormone in hypogonadism, or suppress pathological androgen signalling in androgen-dependent disease.
Androgen deficiency (hypogonadism): Primary hypogonadism (Klinefelter's syndrome, bilateral orchidectomy, cryptorchidism) — LH/FSH elevated, testosterone low. Secondary hypogonadism (pituitary tumour, Kallmann syndrome) — LH/FSH low, testosterone low. Goals: restore serum testosterone to normal range (300–1000 ng/dL), normalise libido and sexual function, maintain bone density and muscle mass, support erythropoiesis. Delayed puberty — short-course testosterone to initiate puberty in constitutional delay.
Androgen excess / androgen-dependent disease:
- Prostate cancer (hormone-sensitive): androgen deprivation to suppress testosterone to castrate levels (<50 ng/dL). Pharmacological castration via: (a) GnRH agonists (leuprolide/goserelin — already covered PH7.4), (b) GnRH antagonists (degarelix), (c) oral anti-androgens (bicalutamide, enzalutamide), (d) CYP17 inhibitors (abiraterone — blocks androgen synthesis adrenal/intra-tumoral).
- Benign prostatic hyperplasia (BPH): DHT drives prostatic epithelial growth. 5α-reductase inhibitors (finasteride 5 mg, dutasteride 0.5 mg) reduce prostate volume — useful in symptomatic BPH with large prostate; effect takes 3–6 months.
- Androgenetic alopecia: finasteride 1 mg (Propecia) — reduces DHT in scalp, slows hair loss in men. Effect takes 3–12 months; must continue indefinitely.
- Hirsutism in women: anti-androgens (cyproterone acetate, spironolactone) or finasteride.
- Precocious puberty (peripheral/adrenal): anti-androgens or adrenal enzyme inhibitors.
SELF-CHECK
Finasteride 5 mg is prescribed for a 65-year-old man with BPH. His PSA is 4.2 ng/mL at baseline. After 12 months on finasteride, PSA is 2.1 ng/mL. How should this be interpreted?
A. A. PSA has halved — this is reassuring and no further prostate cancer workup is needed
B. B. PSA has halved as expected on finasteride (reduces PSA by ~50%); the corrected PSA equivalent is 4.2 ng/mL — prostate cancer workup should proceed if indicated
C. C. PSA reduction indicates the prostate cancer has responded to treatment
D. D. Finasteride should be stopped since it is masking PSA levels
Reveal Answer
Answer: B. B. PSA has halved as expected on finasteride (reduces PSA by ~50%); the corrected PSA equivalent is 4.2 ng/mL — prostate cancer workup should proceed if indicated
Finasteride reduces serum PSA by approximately 50% by reducing prostate epithelial cell mass (DHT-dependent PSA secretion). This is a predictable pharmacological effect — NOT a sign of cancer regression. When interpreting PSA in a man on finasteride, the measured PSA should be doubled to estimate the true equivalent. A PSA that does not fall, or rises, on finasteride is clinically concerning. Stopping finasteride is not indicated — the dosing is correct and the PSA effect is expected. The key clinical message: always document baseline PSA before starting finasteride and inform labs.
Androgen Drugs: Testosterone Preparations, Anabolic Steroids, and Anti-Androgens
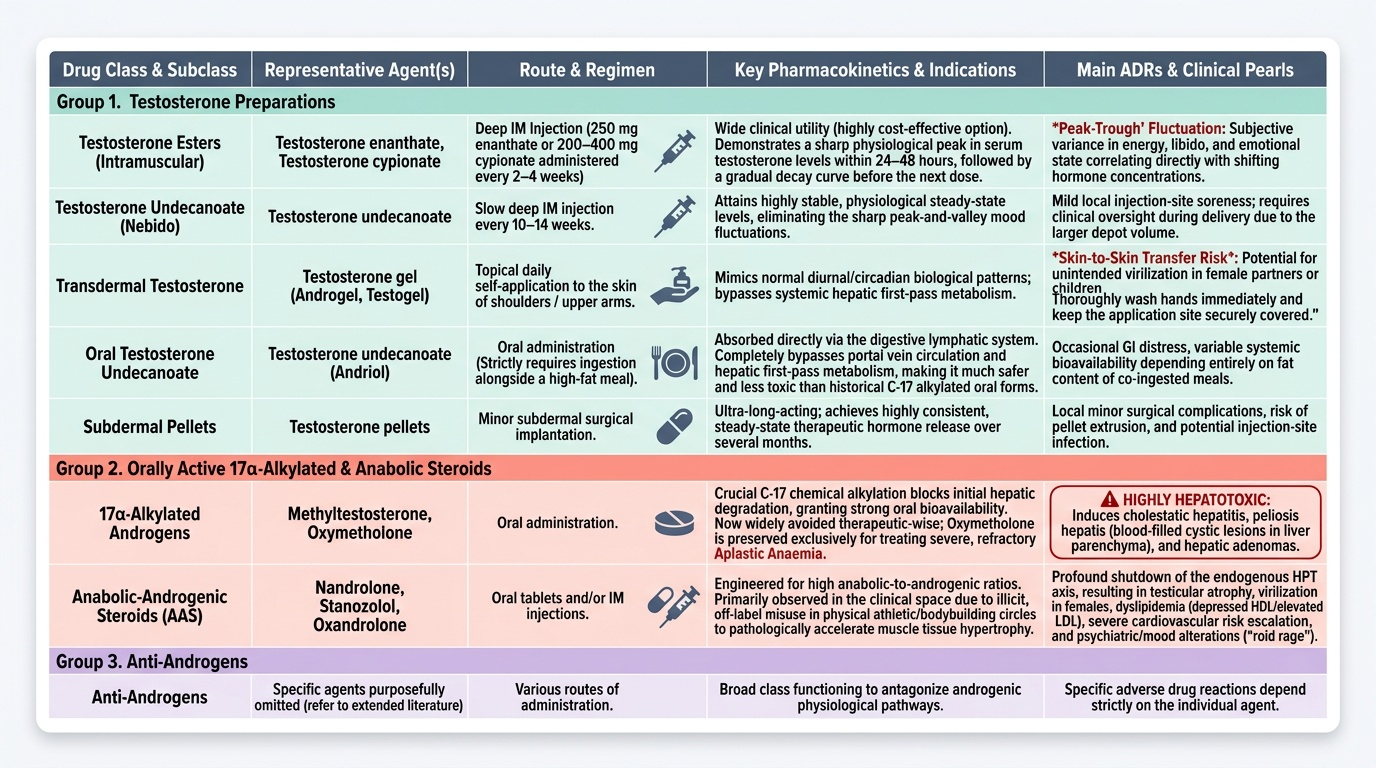
Testosterone preparations differ primarily in route of administration and pharmacokinetic profile — all ultimately provide physiological testosterone.
Testosterone preparations by route:
- IM injections — esters: Testosterone enanthate (250 mg IM every 2–4 weeks), testosterone cypionate (200–400 mg IM every 2–4 weeks) — both medium-acting esters; peak serum T at ~24–48 hours, levels trough before next injection ('peak-trough' pattern). Testosterone undecanoate (Nebido) — very long-acting IM, every 10–14 weeks; more stable levels. IM injections are widely used in India for cost-effectiveness.
- Transdermal: Testosterone gel (Androgel, Testogel) — applied daily to shoulders/upper arms; mimics circadian T pattern; avoids first-pass metabolism; risk of transfer to partners/children via skin contact (counsel accordingly).
- Oral: Testosterone undecanoate (Andriol) — absorbed via lymphatics (bypasses hepatic first-pass); requires fatty meal for absorption; less hepatotoxic than 17α-alkylated forms.
- Subdermal implants: Testosterone pellets — long-acting, consistent levels.
17α-Alkylated Androgens (oral) — HEPATOTOXIC: Methyltestosterone, oxymetholone — orally active due to C-17 alkylation that resists hepatic first-pass but causes cholestatic hepatitis and peliosis hepatis. Now largely avoided for therapeutic use; oxymetholone still used in aplastic anaemia.
Anabolic Steroids (misuse): Nandrolone, stanozolol, oxandrolone — primarily used for muscle mass and endurance (sports doping). ADRs of anabolic steroid abuse: acne, alopecia, gynaecomastia (aromatase conversion), testicular atrophy (suppressed LH/FSH → impaired spermatogenesis), hepatotoxicity (17-alkylated forms), increased RBC (polycythaemia), aggression, libido changes, virilisation in women, premature epiphyseal closure in adolescents, increased LDL/decreased HDL (cardiovascular risk).
5α-Reductase Inhibitors: Finasteride (type 2 isoenzyme inhibitor) and dutasteride (type 1 + type 2 — more complete DHT suppression). Both reduce prostate volume ~20–30% after 6 months; reduce PSA by ~50%. ADRs: erectile dysfunction, decreased libido, ejaculatory dysfunction, gynaecomastia (reduced DHT → relative androgen/oestrogen imbalance). Sexual ADRs may persist after drug cessation (post-finasteride syndrome — controversial).
Anti-Androgens: Cyproterone acetate — AR antagonist AND progestogen; used for hirsutism in women (low dose), severe hypersexuality, prostate cancer (flare protection). Bicalutamide — pure non-steroidal AR antagonist; used in prostate cancer (combined with GnRH agonist or monotherapy); ADR: gynaecomastia (AR block → relative oestrogen excess). Spironolactone — aldosterone antagonist with anti-androgen properties (used for hirsutism and PCOS in women).
Provided image