Page 7 of 25
PH1.6 | PH1.6 | Pharmacokinetics Across ADME — SDL Guide — SDL Guide
Learning Objectives
- Describe the processes of drug absorption, distribution, metabolism, and excretion (ADME) and the key factors influencing each.
- Explain pH-partition theory, ionisation, and first-pass metabolism as determinants of oral bioavailability.
- Define and calculate pharmacokinetic parameters: half-life (t½), volume of distribution (Vd), clearance (CL), steady-state concentration (Css), loading dose, and maintenance dose.
- Distinguish zero-order from first-order elimination kinetics and explain the clinical consequences of each.
- Explain drug metabolism (Phase I/II), enzyme induction, and enzyme inhibition and their impact on plasma drug levels.
INSTRUCTIONS
Pharmacokinetics (PK) is the quantitative description of how a drug moves through the body over time. It answers the practical questions that every prescriber faces: Why does digoxin need a loading dose? Why must aminoglycosides be dosed differently in patients with renal failure? Why does rifampicin make oral contraceptives fail? Understanding ADME — absorption, distribution, metabolism, and excretion — converts these questions from mysteries into predictable, manageable problems. This SDL builds the PK framework that underpins rational dose adjustment across your entire clinical career.
References
- Tripathi KD. Essentials of Medical Pharmacology, 8th ed., Ch 1 (Pharmacokinetics) (textbook)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, 13th ed., Ch 2 (Pharmacokinetics) (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 72-year-old woman with atrial fibrillation is admitted with nausea, vomiting, and visual halos. Her digoxin level is 3.8 ng/mL — nearly double the upper limit of the therapeutic range (0.5–2.0 ng/mL). She weighs 42 kg and has a serum creatinine of 2.8 mg/dL. Her GP had prescribed the standard dose without adjusting for her reduced renal function or small body mass. Digoxin is 60–80% excreted unchanged by the kidneys and has a large volume of distribution that correlates with lean muscle mass. In this frail, renally impaired woman, both elimination and distribution changed dramatically — and no one recalculated the dose. Understanding pharmacokinetics would have prevented this toxicity.
WHY THIS MATTERS
Pharmacokinetics is the scientific basis for individualised dosing. The average dose in a prescribing guideline was derived from population PK studies — it is the dose that works for the average patient. But your patient is rarely the average: they may have renal impairment that slows excretion, hepatic disease that impairs metabolism, extremes of age that alter distribution and clearance, obesity that changes Vd, or genetic polymorphisms in CYP enzymes that make them a rapid or poor metaboliser. Without a working knowledge of ADME and the key PK parameters, you cannot adjust doses rationally — you are guessing. Therapeutic drug monitoring (TDM) for drugs like digoxin, lithium, aminoglycosides, and anti-epileptics depends entirely on your ability to interpret plasma concentrations in the context of PK principles.
RECALL
From Year-1 biochemistry, you know that lipophilic molecules diffuse freely across phospholipid bilayer membranes, while ionised or hydrophilic molecules cannot. You also know that the liver's CYP450 enzyme system oxidises xenobiotics, and that the Henderson-Hasselbalch equation relates pH, pKa, and the ionisation state of a weak acid or base. From physiology, you know that the kidney filters plasma at the glomerulus, secretes some molecules actively into the tubule, and reabsorbs others. These three sets of knowledge — membrane permeability, hepatic enzyme chemistry, and renal handling — are the building blocks of pharmacokinetics.
Why Drug Concentrations Follow Predictable Patterns
Every time a drug is administered, it undergoes a characteristic journey through the body — absorbed from its site of administration, distributed into tissues, transformed by metabolic enzymes, and eventually eliminated via excretion. This journey follows quantifiable, predictable patterns governed by the physical and chemical properties of the drug and the physiological characteristics of the patient. Pharmacokinetics is the branch of pharmacology that describes these patterns mathematically, allowing clinicians to predict drug concentrations at any point in time after a given dose.
The clinical importance of predictable concentration patterns cannot be overstated. For most drugs, therapeutic effect correlates with the concentration of drug at the receptor — too low and the drug is ineffective, too high and it is toxic. The zone between the minimum effective concentration (MEC) and the minimum toxic concentration (MTC) is called the therapeutic window. Pharmacokinetic principles allow prescribers to design dosing regimens that keep plasma concentrations within this window throughout the dosing interval.
Consider the digoxin case from the hook: the standard adult dose of 0.25 mg daily assumes average renal function and average body composition. When renal function falls by 60%, digoxin elimination falls by a proportional amount, and the drug accumulates to toxic levels. This is not a mystery — it is a quantitatively predictable consequence of PK principles that a clinician equipped with the right knowledge can anticipate and prevent. The four phases of a drug's journey — absorption, distribution, metabolism, and excretion (ADME) — each contribute to determining where drug concentrations land within or outside the therapeutic window.
Goals of Pharmacokinetic Understanding in Clinical Practice
Understanding pharmacokinetics serves four immediately practical clinical goals, each of which maps onto a decision you will make as a prescribing clinician. Recognising these goals makes the abstract algebra of PK feel purposeful rather than arbitrary.
The first goal is maintaining concentrations within the therapeutic window. By understanding half-life and dosing interval, the clinician can design a regimen where the trough concentration (just before the next dose) never falls below the MEC, and the peak concentration (just after absorption is complete) never exceeds the MTC. Antibiotics with concentration-dependent killing (aminoglycosides) are dosed to achieve a high peak:MIC ratio; antibiotics with time-dependent killing (beta-lactams) are dosed to maximise the time that concentrations exceed the MIC — two completely different dosing strategies derived from PK-PD integration.
The second goal is dose adjustment in organ impairment. Renal impairment reduces drug excretion; hepatic impairment reduces drug metabolism. A clinician who knows a drug's primary elimination pathway can predict which patients need dose reduction and by how much, using published nomograms or adjusted Cockcroft-Gault creatinine clearance calculations.
The third goal is therapeutic drug monitoring (TDM). For drugs with a narrow therapeutic window and high interpatient PK variability — digoxin, lithium, phenytoin, vancomycin, aminoglycosides, cyclosporine — plasma concentrations are measured and used to individualise dose. TDM is only interpretable when the clinician understands when during the dosing interval to take the sample (trough for aminoglycosides, trough or peak depending on drug), what steady state means, and how many half-lives must elapse before a new steady state is reached after a dose change.
The fourth goal is predicting and managing drug interactions. CYP450 enzyme induction and inhibition alter the metabolism of co-administered drugs in quantitatively predictable ways. A clinician who knows that rifampicin is a potent CYP3A4 inducer and that most oral contraceptives are metabolised by CYP3A4 can predict the interaction and counsel the patient before contraceptive failure occurs.
Absorption and Bioavailability
Absorption is the process by which a drug moves from its site of administration into the systemic circulation. For oral drugs, this means crossing the gastrointestinal epithelium — a selective barrier of columnar epithelial cells joined by tight junctions, spanning a surface area of approximately 200 m² including villi and microvilli. For injected drugs, it means moving from the injection site into capillaries or lymphatics. The rate and extent of absorption together determine the bioavailability of the drug and the shape of its plasma concentration-time curve — specifically the Tmax (time to peak concentration) and Cmax (peak concentration).
Absorption is not a passive mechanical process; it depends on the physicochemical properties of the drug molecule interacting with the biological characteristics of the absorption site. The gastrointestinal tract presents different pH environments along its length (stomach pH 1–3, duodenum pH 5–6, ileum pH 7–8), different transport protein expressions, different blood flow rates, and different surface areas. A drug's behaviour in this varying environment is determined primarily by two intrinsic properties that together govern the ability to cross membranes:
The primary mechanism of absorption across biological membranes is passive diffusion of the non-ionised, lipophilic form of the drug down its concentration gradient. This is governed by those two key properties:
- Lipophilicity: more lipophilic drugs diffuse more readily through phospholipid bilayers. Polar (hydrophilic) drugs cross membranes poorly without transport proteins.
- Ionisation state: most drugs are weak acids or weak bases. The ionised (charged) form is water-soluble but cannot cross membranes; only the non-ionised form diffuses freely. The ratio of ionised to non-ionised drug is determined by the Henderson-Hasselbalch equation and depends on the drug's pKa and the local pH.
pH-partition theory predicts drug distribution across pH compartments. For a weak acid (e.g., aspirin, pKa ≈ 3.5): in the stomach (pH ≈ 2), the acidic environment exceeds the pKa, pushing the equilibrium towards the non-ionised form — aspirin is well absorbed from the stomach. In plasma (pH 7.4), the equilibrium shifts dramatically towards ionised form — aspirin is 'trapped' in plasma as the ionised species. This principle also underpins manipulation of urine pH to accelerate drug excretion (alkalinisation for aspirin overdose).
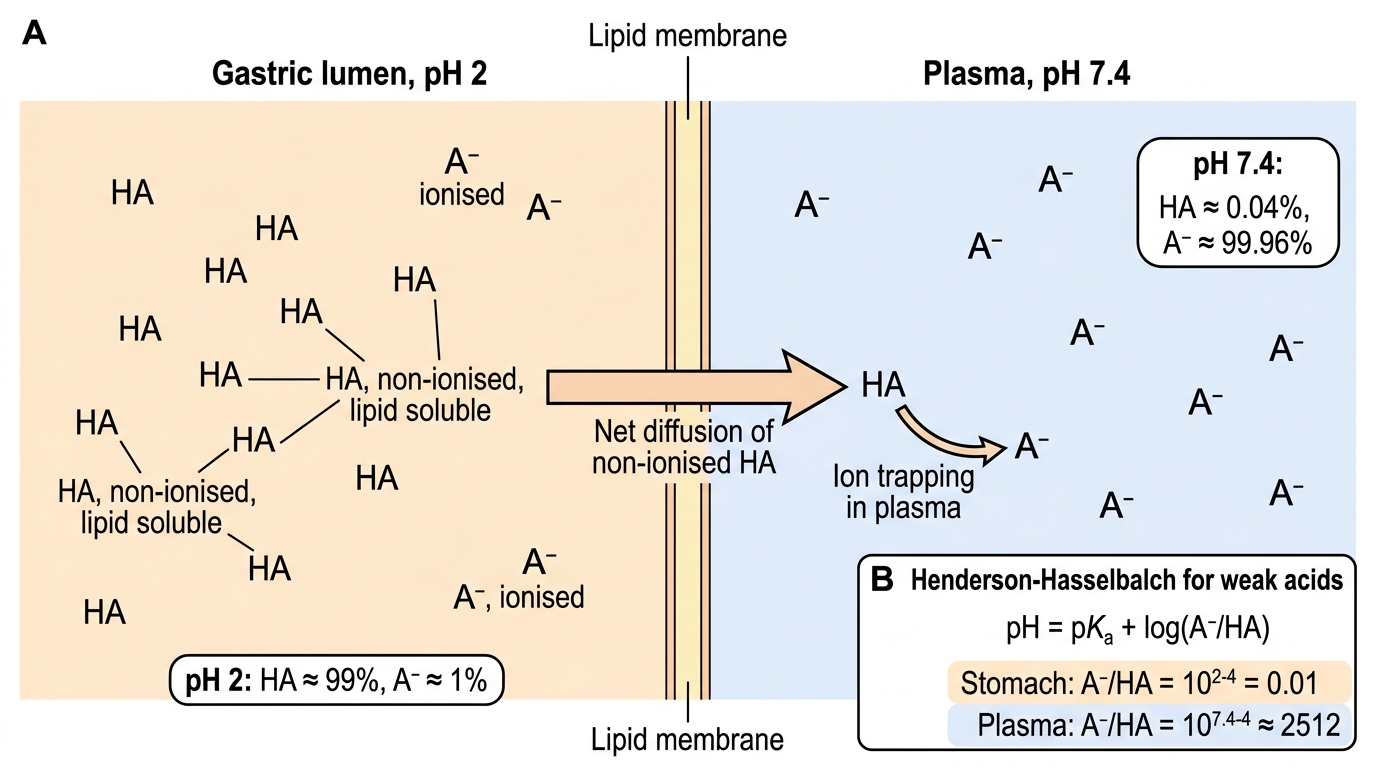
pH-Partition Theory for a Weak Acid Drug
Active transport carries drugs against concentration gradients using membrane transporter proteins (e.g., P-glycoprotein, organic anion transporters). Some drugs are substrates for efflux transporters (like P-gp) that pump them back into the gut lumen, reducing absorption.
Bioavailability (F) is the fraction of an administered dose that reaches the systemic circulation in its active form. For IV drugs, F = 1 (100%) by definition. For oral drugs, F < 1 due to: (a) incomplete absorption from the GI tract, and (b) first-pass hepatic metabolism — the portal blood drains the gut directly into the liver, where hepatic enzymes may extract and metabolise a substantial fraction of the absorbed drug before it reaches systemic circulation. Drugs with high first-pass extraction (e.g., morphine F ≈ 25%, lidocaine F ≈ 35%, GTN F ≈ 1–10%) require parenteral or non-oral mucosal routes, or substantially higher oral doses to compensate.
SELF-CHECK
Aspirin is a weak acid with pKa ≈ 3.5. In the stomach (pH 2), which form predominates, and why does this affect absorption?
A. Ionised form predominates in the stomach; ionised drugs cross membranes more readily.
B. Non-ionised form predominates in the stomach (pH < pKa); non-ionised lipophilic drugs diffuse freely across membranes — so aspirin is absorbed well from the gastric mucosa.
C. Both forms are equal in the stomach at pH 2 (because pH equals pKa at the 50/50 point).
D. Aspirin is always ionised regardless of pH because it is an acid.
Reveal Answer
Answer: B. Non-ionised form predominates in the stomach (pH < pKa); non-ionised lipophilic drugs diffuse freely across membranes — so aspirin is absorbed well from the gastric mucosa.
For a weak acid, when pH < pKa, the non-ionised (HA) form predominates. Stomach pH ≈ 2 < pKa 3.5, so aspirin is predominantly non-ionised in the stomach. Non-ionised molecules are lipophilic and cross phospholipid membranes by passive diffusion — hence aspirin is absorbed from the gastric mucosa. In plasma (pH 7.4 >> pKa 3.5), aspirin is predominantly ionised and 'trapped' in plasma. This is the basis of pH-partition theory.