Page 8 of 25
PH1.6 | PH1.6 | Pharmacokinetics Across ADME — SDL Guide — SDL Guide (Part 2)
Distribution: Volume of Distribution and Protein Binding
Once a drug reaches the systemic circulation, it distributes into tissues at a rate and extent determined by its physical properties and the characteristics of each tissue compartment. Distribution encompasses the reversible transfer of drug from the bloodstream into the various fluid and tissue compartments of the body.
Plasma protein binding is the first determinant of distribution. Most drugs circulate partially bound to plasma proteins — primarily albumin (for acidic drugs and many neutral drugs) and alpha-1 acid glycoprotein (for basic drugs). Bound drug is pharmacologically inactive (cannot cross membranes or bind receptors) and too large for glomerular filtration. Only the free (unbound) fraction is pharmacologically active and available for distribution, receptor binding, metabolism, and excretion. Protein binding is usually expressed as the percentage bound (e.g., warfarin is 99% protein-bound; only 1% is free and active). When two highly protein-bound drugs compete for the same binding sites, displacement interactions can transiently increase the free fraction of the displaced drug — a clinically important drug interaction mechanism.
The volume of distribution (Vd) is a pharmacokinetic parameter that quantifies how extensively a drug distributes into tissues relative to plasma. It is calculated as:
Vd = Dose / Initial plasma concentration
Vd is not a real anatomical volume — it is an apparent volume, defined as the volume in which the drug would need to be dissolved to explain the measured plasma concentration. A small Vd (~5 L = roughly plasma volume) suggests the drug stays mainly in plasma (e.g., warfarin, despite high protein binding — displacement from protein forces it into plasma, keeping Vd small). A large Vd (hundreds of litres — exceeding total body water of ~42 L in an adult) indicates extensive tissue binding; the drug has left the plasma and concentrated in peripheral tissues (e.g., chloroquine Vd ~250–800 L/kg; amiodarone Vd ~60 L/kg). A large Vd has two important clinical consequences: (1) a large loading dose is needed to rapidly achieve therapeutic plasma concentrations, and (2) dialysis is ineffective at removing the drug in overdose (most drug is in tissues, not plasma).
Physiological barriers restrict distribution. The blood-brain barrier (BBB) — formed by tight junctions between cerebral capillary endothelial cells and supported by astrocyte foot processes — permits only lipophilic, non-ionised drugs of low molecular weight to enter the CNS. This protects the brain from many toxins but also means that CNS drug therapy requires specially designed lipophilic compounds (e.g., levodopa, not dopamine, for Parkinson's disease). The placental barrier is less selective than the BBB — most drugs cross to varying degrees, which is why all drug use in pregnancy requires careful risk-benefit assessment.
Metabolism: Phase I, Phase II, Induction, and Inhibition
Drug metabolism (biotransformation) is the enzymatic conversion of a drug to one or more metabolites. Its primary purpose is to convert lipophilic drugs (which would otherwise be reabsorbed from renal tubules) into more polar, water-soluble compounds that can be excreted in urine or bile. The liver is the principal site of drug metabolism, although the gut wall, lung, kidney, and plasma also contribute.
Metabolism is classically divided into two phases, which often (but not always) occur sequentially:
Phase I reactions are functionalisation reactions that add or expose a functional group (-OH, -NH2, -COOH, -SH) on the drug molecule. The primary enzyme system is the cytochrome P450 (CYP450) superfamily — a group of haem-containing monooxygenases located mainly in hepatocyte endoplasmic reticulum. Major CYP isoforms relevant to drug metabolism include CYP3A4 (metabolises ~50% of all drugs — e.g., statins, benzodiazepines, macrolides, many antiretrovirals), CYP2D6 (codeine, tramadol, many antidepressants, beta-blockers), CYP2C9 (warfarin, NSAIDs, phenytoin), and CYP1A2 (theophylline, clozapine, caffeine). Phase I products may be active, inactive, or toxic (the last is the mechanism of paracetamol hepatotoxicity via NAPQI formation by CYP2E1).
Phase II reactions are conjugation reactions that attach a large polar molecule (glucuronate, sulfate, acetyl group, methyl group, or amino acid) to the drug or Phase I metabolite. The resulting conjugate is almost always pharmacologically inactive and highly water-soluble, making it readily excreted by the kidney or in bile. Glucuronidation (UDP-glucuronosyltransferase — UGT enzymes) is the most common Phase II reaction in humans.
Prodrugs are pharmacologically inactive parent compounds that are converted to their active form by metabolism. This strategy is used when the active form is poorly absorbed (enalapril → active enalaprilat; oral enalapril is well absorbed), chemically unstable, or too polar to penetrate membranes. Codeine is a prodrug converted to morphine by CYP2D6; poor metabolisers (CYP2D6 deficient) get no analgesia from codeine, while ultra-rapid metabolisers may develop life-threatening morphine toxicity from a standard codeine dose — a genetic pharmacokinetic interaction.
Enzyme induction occurs when a drug, dietary compound, or environmental chemical increases the expression of CYP450 enzymes by activating nuclear receptors (PXR, CAR) that transcribe more enzyme protein. More enzyme = faster metabolism of substrate drugs = lower plasma levels = reduced therapeutic effect. Clinically important inducers: rifampicin (potent inducer of CYP3A4, CYP2C9, P-glycoprotein — reduces levels of warfarin, oral contraceptives, HIV protease inhibitors, cyclosporine, digoxin); carbamazepine, phenytoin, phenobarbitone; St John's Wort (herbal — frequently overlooked).
Enzyme inhibition directly blocks CYP450 enzyme activity, reducing the metabolism of substrate drugs and increasing their plasma concentrations — potentially to toxic levels. Competitive inhibitors share the same binding site; non-competitive inhibitors bind elsewhere. Clinically important inhibitors: erythromycin and clarithromycin (CYP3A4 inhibitors — increase levels of statins → rhabdomyolysis risk; increase carbamazepine levels); fluconazole (CYP2C9 inhibitor — potentiates warfarin); grapefruit juice (CYP3A4 inhibitor in gut wall — increases levels of statins, nifedipine, cyclosporine — not a drug but a frequent oversight).
| Phase | Reaction types | Key enzymes | Products | Examples |
|---|---|---|---|---|
| Phase I | Oxidation, reduction, hydrolysis | CYP450 (CYP3A4, 2D6, 2C9, 1A2), esterases, amidases | Functionalised metabolites (may be active or inactive) | Paracetamol → NAPQI (CYP2E1); codeine → morphine (CYP2D6) |
| Phase II | Glucuronidation, sulfation, acetylation, methylation | UGT, SULT, NAT, COMT | Water-soluble conjugates (usually inactive) | Morphine → morphine-6-glucuronide (active); isoniazid acetylation (NAT2) |
SELF-CHECK
A patient on warfarin (metabolised by CYP2C9) is started on rifampicin for pulmonary tuberculosis. What is the expected effect on warfarin plasma levels and anticoagulation?
A. Rifampicin inhibits CYP2C9, increasing warfarin levels and anticoagulation effect — risk of bleeding.
B. Rifampicin induces CYP2C9, increasing warfarin metabolism and reducing plasma levels — risk of subtherapeutic anticoagulation and thrombosis.
C. Rifampicin competes with warfarin for protein binding, transiently increasing free warfarin — requires dose reduction.
D. No pharmacokinetic interaction exists between rifampicin and warfarin.
Reveal Answer
Answer: B. Rifampicin induces CYP2C9, increasing warfarin metabolism and reducing plasma levels — risk of subtherapeutic anticoagulation and thrombosis.
Rifampicin is one of the most potent known enzyme inducers, activating PXR and CAR nuclear receptors to transcribe more CYP450 enzymes including CYP2C9 and CYP3A4. Increased CYP2C9 activity accelerates warfarin metabolism, reducing warfarin plasma levels substantially (sometimes by >50%). The clinical consequence is loss of anticoagulant effect — a serious risk of thromboembolic events. Warfarin doses often need to be increased 2-3 fold during rifampicin co-administration, and readjusted downward when rifampicin is stopped (induction reverses within 2-4 weeks). INR must be monitored closely throughout.
Excretion and Key PK Parameters
Drug excretion is the irreversible removal of drug or metabolites from the body — the final phase of the ADME sequence that terminates drug action and prevents accumulation to toxic levels. The kidney is the primary organ of excretion for water-soluble drugs and metabolites; the liver/biliary system handles high-molecular-weight compounds and conjugated metabolites; the lungs excrete volatile anaesthetics and alcohol vapour; and minor pathways include sweat, saliva, and breast milk (the last being clinically important for drug safety in breastfed infants).
Renal excretion is the most quantitatively important pathway for the majority of drugs used in clinical practice. Its efficiency is directly coupled to glomerular filtration rate (GFR), which in turn is determined by renal perfusion, glomerular membrane integrity, and tubular function. Any disease state that reduces GFR — chronic kidney disease, acute kidney injury, haemodynamic compromise — proportionally reduces the renal excretion of drugs cleared primarily by this route. This is why dose adjustment is mandatory for drugs such as aminoglycosides, digoxin, vancomycin, metformin, and most penicillins in patients with renal impairment. Understanding the three mechanisms of renal drug handling allows the clinician to predict which drugs are affected and by how much. The kidney handles drugs through three distinct nephron processes that together determine net excretion:
Renal excretion involves three processes in the nephron:
1. Glomerular filtration: free (unbound) drug is filtered at the glomerulus at a rate proportional to GFR (~125 mL/min in a healthy adult). Highly protein-bound drugs are not filtered (too large). This is why renal impairment reduces excretion of low-protein-bound drugs like aminoglycosides and digoxin disproportionately.
2. Active tubular secretion: organic anion transporters (OAT) and organic cation transporters (OCT) in the proximal tubule actively secrete drugs into the tubular lumen against concentration gradients. This can clear protein-bound drug efficiently and explains why some drugs are eliminated faster than GFR alone would predict. Drug interactions can occur at this level (probenecid blocks OAT secretion of penicillin — once exploited to prolong penicillin action).
3. Passive tubular reabsorption: non-ionised, lipophilic drugs in the tubular lumen diffuse back across the tubular epithelium into the bloodstream down the concentration gradient, reducing net excretion. pH-dependent excretion exploits this: alkalinising the urine (sodium bicarbonate) ionises weak acids (aspirin, phenobarbitone) in the tubular lumen, trapping them and promoting excretion — the therapeutic basis for alkaline diuresis in aspirin overdose.
Biliary excretion occurs for high-molecular-weight drugs and conjugated metabolites. After biliary secretion into the duodenum, some drugs are re-absorbed from the intestine — this cycle of biliary excretion → intestinal reabsorption → hepatic recycling is called enterohepatic circulation and significantly prolongs the half-life of drugs subject to it (e.g., estrogens, some NSAIDs). Interrupting enterohepatic circulation (e.g., cholestyramine binding estrogens in the gut) accelerates elimination.
Key pharmacokinetic parameters:
- Half-life (t½): the time for plasma drug concentration to fall by 50%. For first-order kinetics: t½ = 0.693 × Vd / CL. A drug takes 4–5 half-lives to reach steady state (after repeated dosing) and 4–5 half-lives to be essentially eliminated after stopping.
- Clearance (CL): the volume of plasma cleared of drug per unit time (mL/min or L/hr). Total clearance = hepatic clearance + renal clearance + other. CL is the primary determinant of steady-state concentration.
- Steady-state concentration (Css): with repeated dosing at a fixed interval, plasma concentrations reach a plateau after 4–5 half-lives where the rate of input equals the rate of elimination. Css = (F × Dose) / (CL × dosing interval). Importantly, steady state is reached after 4–5 t½ regardless of dose size — increasing the dose does not accelerate the time to reach steady state.
- Loading dose (Ld): a large initial dose given to rapidly achieve therapeutic concentrations before steady state: Ld = Vd × target Css / F. Used for drugs with long half-lives (digoxin t½ ~36–48 hr; loading dose achieves effect within hours).
- Maintenance dose (Md): the dose given at each dosing interval to maintain Css: Md = CL × Css × dosing interval / F.
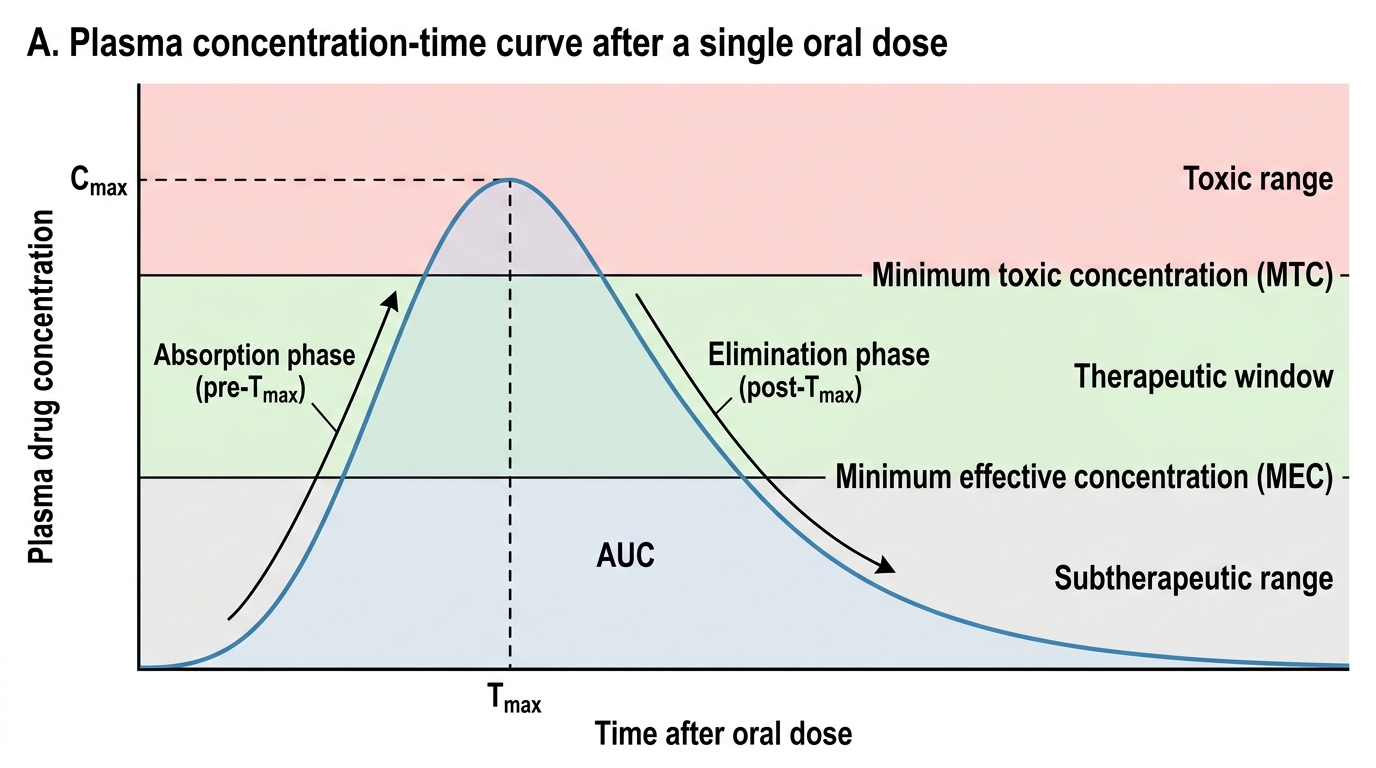
Single Oral Dose Plasma Concentration-Time Curve