Page 5 of 34
PH3.2 | PH3.2 | Sedative and Hypnotic Agents — SDL Guide — SDL Guide (Part 2)
Benzodiazepines — PK, PD, Uses, and ADRs
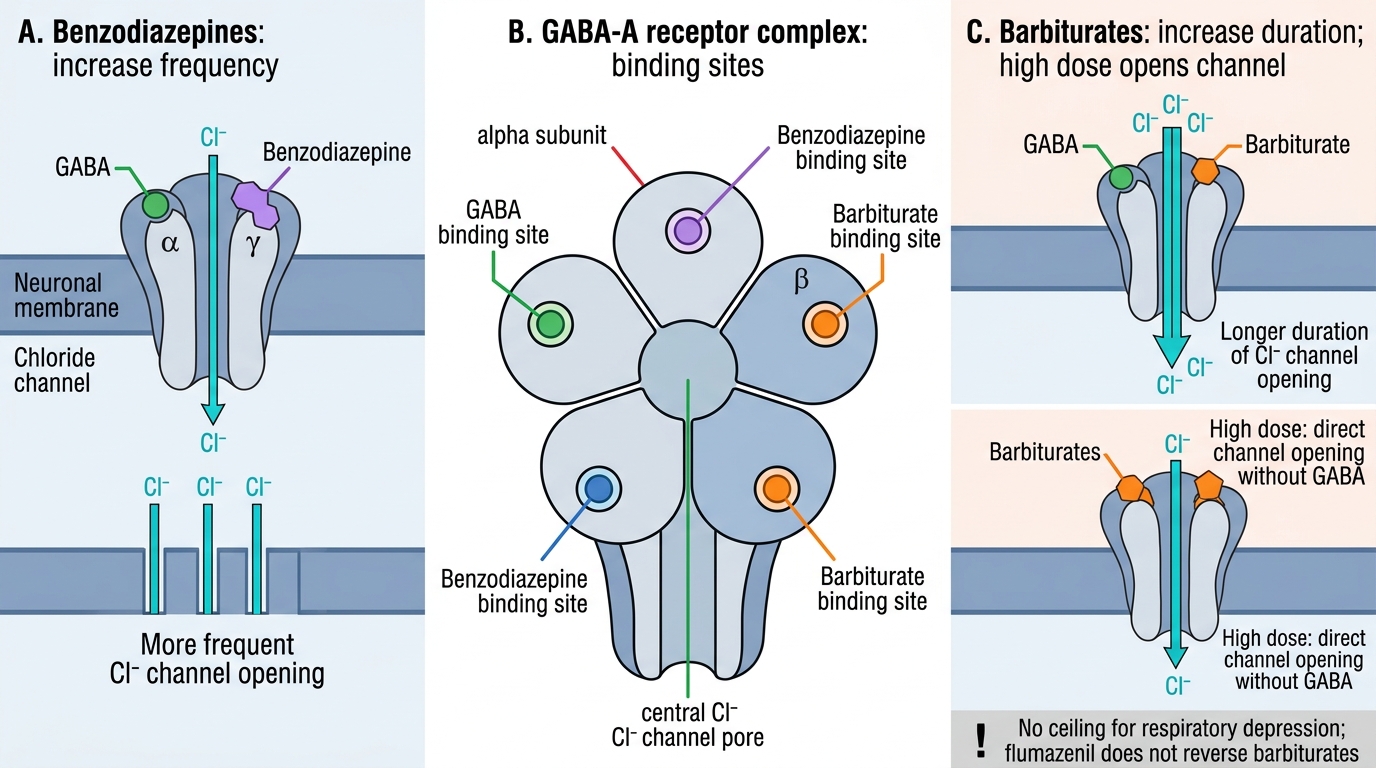
Benzodiazepines (BZDs) bind to a specific allosteric site on the GABA-A receptor (at the α-γ subunit interface) and potentiate GABA's effect by increasing the frequency of Cl⁻ channel opening — they do not open the channel in the absence of GABA. This ceiling effect is why BZDs have a wider therapeutic index than barbiturates. Their pharmacological effects include sedation, anxiolysis, anticonvulsant activity, amnesia, and muscle relaxation — all consequences of enhanced GABAergic inhibition at different brain regions.
Pharmacokinetically, BZDs are classified by duration of action, which depends on lipophilicity, redistribution, and presence of active metabolites. Diazepam is long-acting (t½ 20-100h) partly because its active metabolite desmethyldiazepam (t½ up to 200h) extends the effect — this explains why repeated dosing in elderly patients causes progressive accumulation and prolonged sedation. Lorazepam has no active metabolite and is therefore preferred in hepatic impairment and in elderly patients requiring benzodiazepine therapy (conjugation only, not oxidation). Midazolam is ultra-short-acting (IV) and is preferred for procedural sedation.
| Agent | Duration | Active Metabolite | Key Use |
|---|---|---|---|
| Diazepam | Long (t½ ~40h) | Desmethyldiazepam | Anxiety, SE, muscle spasm, alcohol withdrawal |
| Lorazepam | Intermediate | None | SE (preferred IV in status), pre-op, hepatic impairment |
| Midazolam | Short (IV: ~2h) | Minor | Procedural sedation, pre-op, ICU sedation |
| Temazepam | Short | None | Sleep-onset insomnia |
| Flurazepam | Long | Active (desalkylflurazepam) | Sleep-maintenance (rarely used now) |
Benzodiazepines vs Barbiturates at the GABA-A Receptor
ADRs: daytime sedation, cognitive impairment, anterograde amnesia, ataxia, respiratory depression (especially with opioids or alcohol), tolerance (pharmacodynamic — receptor down-regulation), physical dependence (with use >4 weeks), and withdrawal syndrome (anxiety, insomnia, tremor, seizures — analogous to alcohol withdrawal because both involve GABA-A). Falls and hip fractures are a major risk in elderly patients.
Reversal: flumazenil is a competitive antagonist at the BZD binding site of GABA-A. It reverses BZD-induced sedation and respiratory depression rapidly (onset 1-2 min IV). Caution: short half-life (~1h) means re-sedation occurs if the BZD has a longer duration — may need repeated doses or infusion. Does NOT reverse barbiturate-induced coma.
Barbiturates — PK, PD, Uses, and ADRs
Barbiturates bind to a distinct site on the GABA-A receptor (β-subunit region) and potentiate GABA-A by increasing the duration of Cl⁻ channel opening. Critically, at supra-therapeutic concentrations barbiturates can open the Cl⁻ channel directly without GABA — this explains their dose-dependent respiratory depression without a ceiling, the absence of a specific reversal agent, and their narrow therapeutic index compared to BZDs.
Pharmacokinetically, barbiturates vary widely by duration. Thiopental (ultra-short, IV) is used as a GA induction agent (covered in PH3.1). Phenobarbitone (phenobarbital) is a long-acting barbiturate used primarily as an anticonvulsant (first-line in neonatal seizures, febrile convulsions, and epilepsy in resource-limited settings); its long half-life (80-100h) allows once-daily dosing. Secobarbital and pentobarbital are shorter-acting agents used historically as sedative-hypnotics but now largely obsolete.
A critical pharmacokinetic property of barbiturates is potent CYP450 enzyme induction — phenobarbitone induces CYP1A2, CYP2C9, CYP2C19, and CYP3A4, accelerating the metabolism of warfarin, oral contraceptives, corticosteroids, and many other drugs. This is a major source of drug interactions in patients on polypharmacy.
ADRs: CNS depression (sedation → coma at overdose), respiratory depression (no ceiling effect), paradoxical excitement in elderly and children, enzyme induction (drug interactions), physical dependence, withdrawal seizures, and porphyria precipitation (like thiopental — both induce δ-ALA synthase). Barbiturate overdose has no specific antidote — management is supportive (airway, ventilation, alkalinisation of urine to enhance phenobarbitone excretion).
Z-Drugs and Newer Hypnotics — PK, PD, and Uses
The Z-drugs — zolpidem, zopiclone, and zaleplon — are structurally distinct from benzodiazepines but act at the same BZD binding site of the GABA-A receptor. Their key distinction is relative selectivity for the α₁ subunit of the GABA-A receptor, which mediates sedation and hypnosis, with less activity at α₂ (anxiolysis), α₃ (muscle relaxation), and α₅ (memory) subunits. This selectivity was proposed to produce a cleaner hypnotic effect without the full spectrum of BZD side-effects, though clinically the differences are modest and dependence still occurs.
Zolpidem has rapid onset and short half-life (~2h), making it suitable for sleep-onset insomnia; it is less useful for sleep-maintenance. Zopiclone has a slightly longer action (~5-6h half-life) and is used for both sleep-onset and sleep-maintenance insomnia; patients frequently report a metallic taste (characteristic ADR). Zaleplon has the shortest half-life of the three (~1h), allowing middle-of-night dosing without next-morning impairment.
All Z-drugs carry risks of tolerance, dependence, parasomnias (sleep-walking, sleep-eating, sleep-driving — a unique and clinically important ADR class) and rebound insomnia on withdrawal. They are not safer than BZDs with regard to dependence.
Ramelteon is a selective melatonin MT₁/MT₂ receptor agonist used for circadian-rhythm-related insomnia and sleep-onset delay. No dependence liability, no GABA-A action, not a controlled substance — useful in elderly patients where BZD/Z-drug risk is high. Suvorexant is a dual orexin receptor antagonist (DORA) that blocks the wake-promoting orexin signal; approved for sleep-onset and sleep-maintenance insomnia; no dependence potential comparable to GABA-A agonists.
SELF-CHECK
A patient presents with acute benzodiazepine overdose — drowsy but breathing spontaneously. What is the appropriate pharmacological reversal agent and its mechanism?
A. Naloxone — opioid receptor antagonist that also reverses benzodiazepine sedation
B. Flumazenil — competitive antagonist at the benzodiazepine binding site of the GABA-A receptor
C. Physostigmine — acetylcholinesterase inhibitor that reverses anticholinergic CNS depression
D. Activated charcoal — adsorbs the benzodiazepine in the gut to prevent absorption
Reveal Answer
Answer: B. Flumazenil — competitive antagonist at the benzodiazepine binding site of the GABA-A receptor
Flumazenil is a competitive antagonist at the benzodiazepine binding site on the GABA-A receptor complex. It rapidly reverses BZD-induced sedation and respiratory depression (onset 1-2 min IV). Caution: flumazenil has a short half-life (~1h) — re-sedation can occur if the ingested BZD has a longer duration, requiring repeat dosing. Naloxone reverses opioids, physostigmine reverses anticholinergics, and activated charcoal is a decontamination measure (not a reversal agent).
SELF-CHECK
Which mechanism correctly distinguishes benzodiazepines from barbiturates at the GABA-A receptor?
A. Benzodiazepines increase the DURATION of Cl⁻ channel opening; barbiturates increase the FREQUENCY
B. Benzodiazepines increase the FREQUENCY of Cl⁻ channel opening; barbiturates increase the DURATION (and can open the channel without GABA)
C. Both benzodiazepines and barbiturates act identically — only duration of action differs
D. Benzodiazepines block GABA-A; barbiturates activate it — opposite mechanisms
Reveal Answer
Answer: B. Benzodiazepines increase the FREQUENCY of Cl⁻ channel opening; barbiturates increase the DURATION (and can open the channel without GABA)
Benzodiazepines allosterically potentiate GABA by increasing the FREQUENCY of Cl⁻ channel opening — they require GABA to be present (ceiling effect, hence wider therapeutic index). Barbiturates increase the DURATION of channel opening, and at high doses can open the channel directly without GABA — this eliminates the ceiling effect and explains the dose-dependent respiratory depression and narrow therapeutic index of barbiturates. Both classes enhance GABAergic inhibition but by different mechanisms.
SELF-CHECK
An 80-year-old man with mild liver impairment needs a benzodiazepine for acute anxiety. Which BZD is MOST appropriate and why?
A. Diazepam — the gold standard BZD with the most evidence for anxiety
B. Flurazepam — long duration ensures sustained anxiolysis
C. Lorazepam — intermediate duration, no active metabolites, undergoes conjugation only
D. Midazolam — fastest onset, easily titrated
Reveal Answer
Answer: C. Lorazepam — intermediate duration, no active metabolites, undergoes conjugation only
Lorazepam is preferred in elderly patients with hepatic impairment because it has no active metabolite (diazepam's active metabolite desmethyldiazepam prolongs action significantly in the elderly and accumulates with hepatic impairment) and undergoes Phase II conjugation (glucuronidation) only — not oxidative Phase I metabolism, which is impaired in liver disease and ageing. Flurazepam's active metabolites accumulate even more than diazepam's. Midazolam is appropriate for procedural sedation but has ultra-short action unsuitable for sustained anxiolysis.
CLINICAL PEARL
Benzodiazepine withdrawal can cause seizures — exactly like alcohol withdrawal. Any patient who has been on a BZD daily for >4 weeks should NOT have it abruptly stopped. Taper gradually over 4-8 weeks using a longer-acting agent (diazepam) if a short-acting BZD was used, to prevent withdrawal seizures. Also remember: Z-drugs are not free of dependence risk — the FDA has added boxed warnings after reports of complex sleep behaviours (sleep-walking, sleep-driving) and dependence. Patients should be warned explicitly.