Page 22 of 34
PH3.8 | PH3.8 | Alcohol Toxicity Management — SDL Guide — SDL Guide
Learning Objectives
- Explain the metabolic pathways of ethanol and methanol and identify the toxic metabolite in methanol poisoning
- Describe the pharmacological management of acute methanol poisoning, including fomepizole and ethanol as antidotes
- Describe the pharmacological management of chronic ethanol intoxication, withdrawal, and Wernicke's encephalopathy
- Explain the mechanism of disulfiram and its role in alcohol use disorder
INSTRUCTIONS
Methanol poisoning — from illicit alcohol, contaminated hand sanitizer, or industrial exposures — is a medical emergency in which delayed treatment causes irreversible blindness and death. Chronic ethanol toxicity creates a spectrum from daily hangovers to fatal withdrawal seizures and irreversible brain damage. This module builds the pharmacological framework for identifying and managing both acute methanol poisoning and the complications of chronic alcohol use.
References
- Tripathi KD. Essentials of Medical Pharmacology, 8th ed., Ch 28 (Alcohols) and Ch 4 (Drug Toxicity and Poisoning) (textbook)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, 13th ed., Ch 23 (Alcohol Use Disorders) (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
During a festival in a rural district, 40 people consume what they believe is locally distilled liquor. Within 12-18 hours, ten develop nausea, vomiting, and severe metabolic acidosis. Several complain of visual blurring; two become permanently blind. Three die. The liquor was adulterated with industrial methanol. The tragedy was not inevitable — early recognition, administration of fomepizole (or ethanol) to block further toxic metabolite formation, and haemodialysis to remove existing toxin can save lives. Understanding methanol's pharmacokinetics and why formic acid is the toxic culprit is the pharmacological key to this emergency.
WHY THIS MATTERS
Methanol poisoning from illicit alcohol outbreaks is a recurring public health emergency in India and globally. You will encounter alcohol withdrawal in any hospital ward — the patient who develops seizures on day 2 of admission after withholding alcohol needs benzodiazepine management, not just observation. Thiamine deficiency in an alcoholic patient who receives IV glucose without prior thiamine can precipitate Wernicke's encephalopathy — a preventable tragedy. These are everyday clinical scenarios with pharmacologically correct interventions that save lives.
RECALL
Recall from biochemistry: alcohol dehydrogenase (ADH) in the liver (and gastric mucosa) oxidises primary alcohols to aldehydes. Aldehyde dehydrogenase (ALDH) oxidises aldehydes to organic acids. The key enzyme cofactor is NAD⁺ → NADH. Recall that ethanol is both a GABA-A potentiator (like benzodiazepines and barbiturates — explains its sedative and anticonvulsant properties in acute intoxication) and an NMDA receptor inhibitor (glutamate antagonism — explains its amnestic and neuroprotective intoxication effects). Chronic ethanol induces NMDA upregulation and GABA-A down-regulation — the basis of tolerance and the mechanism of withdrawal hyperexcitability.
Pathophysiology of Alcohol Metabolism and Toxicity
Ethanol is primarily metabolised in the liver by two pathways: (1) ADH → ALDH pathway (primary, low-dose): ethanol → acetaldehyde (by alcohol dehydrogenase) → acetate (by aldehyde dehydrogenase). Acetaldehyde is toxic (responsible for the flush reaction in ALDH-deficient individuals — common in East Asian populations — and for hangover symptoms) but is normally rapidly metabolised. Acetate is ultimately metabolised to CO₂ and water. (2) Microsomal ethanol oxidising system (MEOS/CYP2E1) (high doses, chronic use): induced by chronic ethanol and is the basis for drug interactions (CYP2E1 induction accelerates metabolism of paracetamol → hepatotoxic NAPQI at lower doses than usual).
Methanol is metabolised by the same enzymes but to very different products: methanol → formaldehyde (by ADH) → formic acid (by ALDH). Formic acid (formate) is the primary toxic metabolite, not formaldehyde (which is rapidly further oxidised). Formic acid causes: (1) high anion-gap metabolic acidosis (formate accumulation); (2) optic nerve toxicity (formate selectively inhibits cytochrome c oxidase in the optic nerve mitochondria → optic atrophy → blindness); (3) CNS depression.
Critically: the metabolism of methanol to its toxic products is slow (hours) relative to ethanol — this explains the latent period (6-24h) between methanol ingestion and symptom onset. Concurrent ethanol ingestion delays methanol toxicity even further (ethanol competes with methanol for ADH — the basis of ethanol as an antidote).
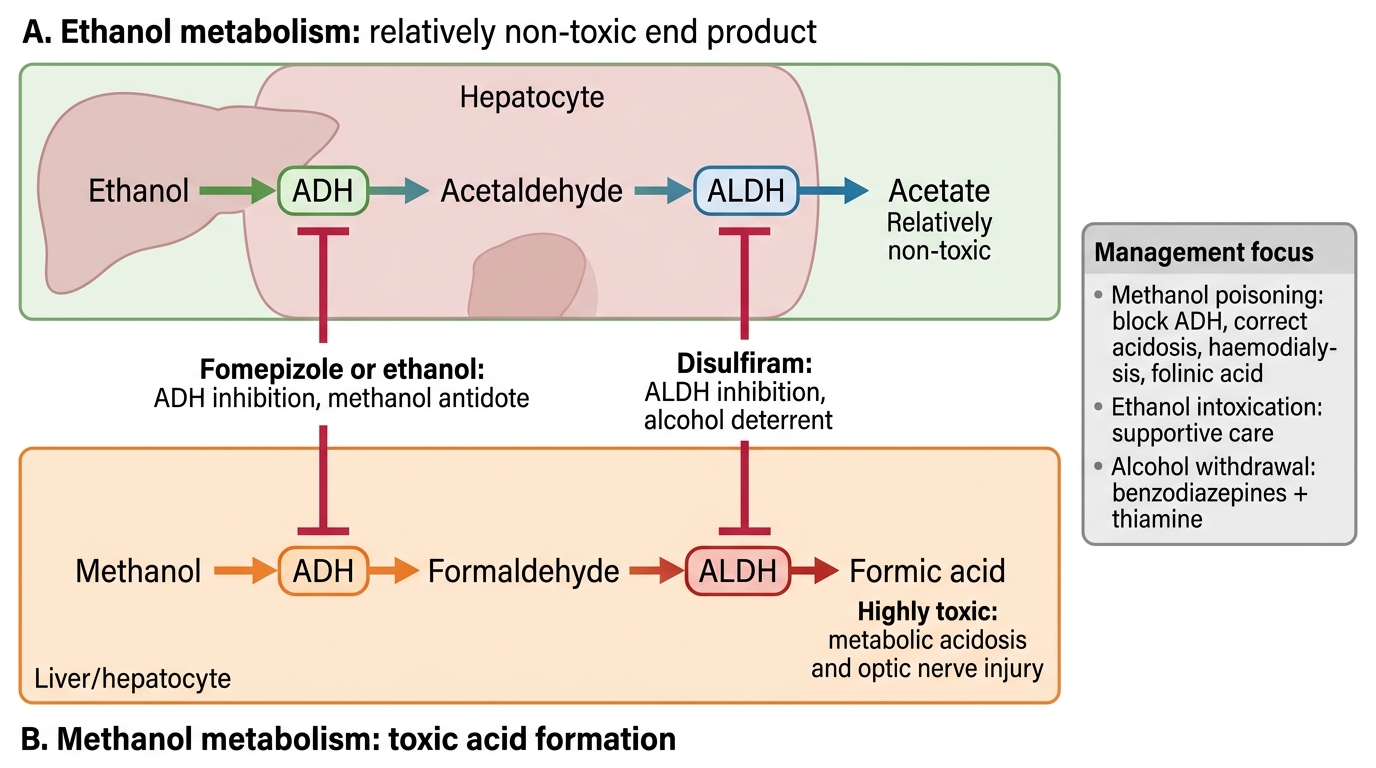
Ethanol vs Methanol Metabolism and Antidote Targets
Goals of Alcohol Toxicity Management
The pharmacological goals differ by the clinical scenario:
Acute methanol poisoning: (1) Prevent further formation of formic acid by blocking ADH (fomepizole or ethanol); (2) Correct the existing metabolic acidosis (sodium bicarbonate); (3) Enhance formate elimination (haemodialysis — also removes methanol directly); (4) Enhance formate metabolism (folinic acid facilitates formate oxidation to CO₂ via tetrahydrofolate pathway).
Acute ethanol intoxication: primarily supportive — airway protection, hydration, blood glucose monitoring (ethanol causes hypoglycaemia by inhibiting hepatic gluconeogenesis). No specific antidote.
Ethanol withdrawal: prevent and treat withdrawal seizures and delirium tremens using long-acting BZDs (diazepam or chlordiazepoxide) via symptom-triggered protocols (CIWA-Ar). Prevent Wernicke's encephalopathy with parenteral thiamine.
Chronic alcohol use disorder — deterrent therapy: disulfiram (ALDH inhibitor) — causes an aversive reaction with any alcohol intake, acting as a pharmacological deterrent. Naltrexone (opioid receptor antagonist) — reduces craving and reward from alcohol (mesolimbic dopamine pathway modulation). Acamprosate (weak NMDA antagonist + GABA-B agonist) — reduces withdrawal symptoms and craving.
The overarching therapeutic principle: treat the immediate life-threatening pathology first (methanol formate; withdrawal seizures; Wernicke's), then address long-term alcohol use disorder.
Classification of Pharmacological Interventions in Alcohol Toxicity
Pharmacological interventions in alcohol toxicity are classified by their mechanism and the toxicity they address:
1. ADH inhibitors (antidotes for methanol/ethylene glycol poisoning):
- Fomepizole (4-methylpyrazole): competitive inhibitor of ADH — first-line antidote for methanol and ethylene glycol poisoning. Blocks conversion of methanol to formaldehyde/formate. IV administration.
- Ethanol: a competitive substrate for ADH with higher affinity than methanol — diverts ADH to metabolise ethanol preferentially, leaving methanol unmetabolised (eliminated slowly by lungs and kidneys). Used when fomepizole is unavailable.
2. ALDH inhibitors (deterrent therapy):
- Disulfiram: irreversible inhibitor of aldehyde dehydrogenase — blocks acetaldehyde metabolism; alcohol ingestion → acetaldehyde accumulation → aversive reaction.
3. BZDs (alcohol withdrawal): diazepam, chlordiazepoxide, lorazepam — treat and prevent alcohol withdrawal seizures and delirium tremens (GABA-A potentiation replaces ethanol's GABA-A effect during detoxification).
4. Vitamins and metabolic support: thiamine (vitamin B1) — prevents and treats Wernicke's encephalopathy; multivitamins; magnesium (often depleted in chronic alcoholism; hypomagnesaemia lowers seizure threshold).
5. Anti-craving agents: naltrexone (μ-opioid antagonist — reduces reward of drinking), acamprosate (NMDA antagonist/GABA-B agonist — reduces post-abstinence craving).